DIAGNOSIS
Clinical Presentation
A general approach to the diagnosis of ILD is presented in Figure 17-1.3
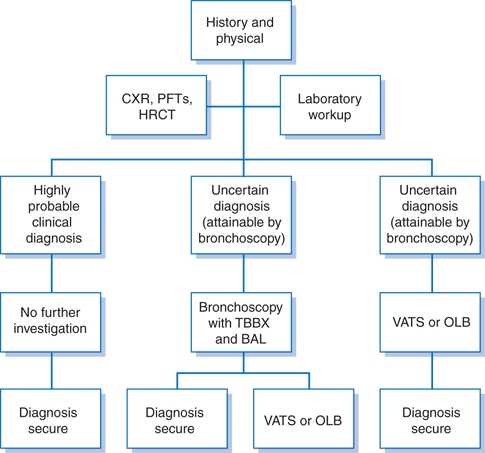
Figure 17-1 Approach to diagnosing interstitial lung diseases. (BAL, bronchoalveolar lavage; CXR, plain chest radiography; HRCT, high-resolution computed tomography; OLB, open lung biopsy; PFTs, pulmonary function tests; TBBX, transbronchial biopsy; VATS, video-assisted thoracoscopic surgery). (Modified from British Thoracic Society. The diagnosis, assessment and treatment of diffuse parenchymal lung disease in adults. Thorax 1999;54:S1−S28.)
History
- History is an extremely important component of the evaluation of ILDs.
- Demographics can be helpful (e.g., lymphangioleiomyomatosis [LAM] is most common in young women). Age of onset of disease is highly variable. IPF typically presents in the seventh decade or later, while CTD-related ILD often presents in the third to fifth decade.
- The temporal course of symptoms should be noted. For example, acute interstitial pneumonia (AIP), cryptogenic organizing pneumonia (COP), acute eosinophilic pneumonia (AEP), and alveolar hemorrhage syndromes develop quite rapidly (days to weeks), whereas IPF, nonspecific interstitial pneumonitis (NSIP), hypersensitivity pneumonitis (HP), and sarcoidosis have a more insidious onset (months to years).
- The most common presenting symptom is dyspnea on exertion. Cough is also common. Nonpulmonary symptoms, such as dysphagia, Raynaud phenomenon, myalgias, and arthralgias, point toward an underlying CTD.
- Active smoking is associated with respiratory bronchiolitis-ILD (RB-ILD), desquamative interstitial pneumonia (DIP), and pulmonary Langerhans cell histiocytosis (PLCH).
- Occupational and environmental exposures can reveal disorders such as HP and pneumoconiosis.
- Medication history (e.g., amiodarone) or treatment with chemotherapy agents and radiation may reveal an underlying cause.
- Family history may also be important (e.g., familial form of IPF).
Physical Examination
- The hallmark feature of ILD is auscultatory crackles; IPF is associated with fine crackles that mimic Velcro ripping apart.
- Clubbing of the nails is an important clue in the diagnosis of IPF.
- Systemic findings of fever, joint pains, and rashes are more indicative of an associated CTD or sarcoidosis.
- Findings of a CTD, including cutaneous telangiectasias, sclerodactyly, arthritis, myositis, and joint deformities, may be present on examination.
- Advanced cases can exhibit findings of pulmonary hypertension and cor pulmonale.
Diagnostic Testing
Laboratory Tests
Laboratory tests should be interpreted in the appropriate clinical setting (Table 17-2).4 Angiotensin-converting enzyme level for sarcoidosis is generally not useful due to its low sensitivity and specificity.
TABLE 17-2 Helpful Blood and Urine Tests to Evaluate ILDs
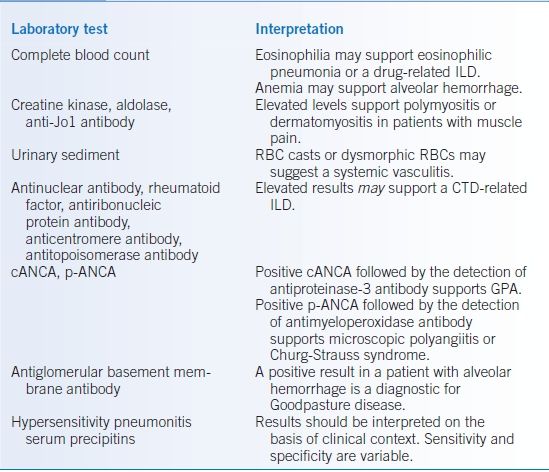
cANCA, cytoplasmic antineutrophilic cytoplasmic antibody; CTD, connective tissue disease; ILD, interstitial lung disease; p-ANCA, perinuclear antineutrophilic cytoplasmic antibody; RBC, red blood cell.
Data from Reynolds H, Matthay R. Diffuse interstitial and alveolar lung diseases. In: George RB, Light RW, Matthay MA, et al. eds. Chest Medicine: Essentials of Pulmonary and Critical Care Medicine. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:262–313.
Imaging
- Plain chest radiography can be helpful in revealing an ILD but not typically diagnostic. CXR can be normal in up to 10% of patients with clinically significant ILDs.4 Radiographic patterns that may be associated with ILDs include the following1,5:
- Small lung volumes: IPF and CTD related
- Preserved or large lung volumes: ILDs associated with airway involvement such as HP, LAM, PLCH, combined pulmonary fibrosis and emphysema (CPFE), and neurofibromatosis-associated ILD
- Upper lobe predominance: sarcoidosis, silicosis, and PLCH
- Lower lobe predominance: IPF and asbestosis
- Peripheral zone predominance: COP and chronic eosinophilic pneumonia (CEP)
- Migratory infiltrates: COP, HP, and eosinophilic pneumonia
- Small lung volumes: IPF and CTD related
- High-resolution computed tomography (HRCT) is the most helpful radiographic test to evaluate ILDs and will also help guide location for lung biopsy and possible mediastinal lymph node sampling. Patterns on HRCT that may be associated with ILDs include the following1,4:
- Reticular lines with honeycombing and traction bronchiectasis: IPF, CTD related, asbestosis, and sarcoidosis
- Nodules: pneumoconiosis, malignancy, rheumatoid arthritis (RA), granulomatosis with polyangiitis (GPA, formerly Wegener granulomatosis), HP, and sarcoidosis (nodular pattern particularly along bronchovascular bundles is common in sarcoidosis)
- Cystic disease: LAM and PLCH
- Honeycombing: IPF, asbestosis, CTD related, chronic HP
- Ground-glass opacities: alveolar hemorrhage, HP, AIP, drug-induced diseases, pulmonary alveolar proteinosis (PAP), NSIP
- Multifocal consolidation: COP or secondary bronchiolitis obliterans organizing pneumonia (BOOP)
- Hilar/mediastinal lymphadenopathy: sarcoidosis, berylliosis, silicosis
- Crazy paving: PAP
- Reticular lines with honeycombing and traction bronchiectasis: IPF, CTD related, asbestosis, and sarcoidosis
Diagnostic Procedures
- Pulmonary function tests (PFTs) should include spirometry, lung volumes, diffusion capacity, and exercise oximetry as well as an arterial blood gas in certain circumstances.
- PFTs classically show a restrictive ventilatory defect: low total lung capacity, low vital capacity, low forced expiratory volume in 1 second (FEV1), and low forced vital capacity (FVC) but normal or high FEV1/FVC.5
- Preserved lung volumes in the setting of ILD should raise suspicion for HP, PLCH, LAM, or CPFE.
- An obstructive ventilatory defect in the setting of ILD could indicate sarcoidosis, HP, PLCH, or LAM due to concomitant airway involvement.
- Abnormal gas exchange is often evident with a low diffusion capacity for carbon monoxide (DLCO) and hypoxemia, particularly with exercise. Elevated DLCO is consistent with alveolar hemorrhage.
- PFTs classically show a restrictive ventilatory defect: low total lung capacity, low vital capacity, low forced expiratory volume in 1 second (FEV1), and low forced vital capacity (FVC) but normal or high FEV1/FVC.5
- Lung biopsy can provide a firm histopathologic diagnosis allowing a clinician to more accurately predict prognosis, assess disease activity, and exclude neoplastic or infectious processes that may mimic an ILD.
- In IPF, a biopsy is not always required as the aforementioned clinical and radiographic criteria can accurately predict the diagnosis in >90% of cases (Table 17-3).6
- There are three routes for lung biopsies: transbronchial lung biopsy via bronchoscopy, wedge biopsy via open thoracotomy or open lung biopsy, and biopsy via video-assisted thoracoscopic surgery (VATS).2 ILD pathology is frequently inhomogeneous. Thus, large specimens typically >2 cm in diameter from more than one lobe of the lung are ideal.
- There are six idiopathic interstitial pneumonias classified on the basis of histopathology (Table 17-4).2,6
- In IPF, a biopsy is not always required as the aforementioned clinical and radiographic criteria can accurately predict the diagnosis in >90% of cases (Table 17-3).6
TABLE 17-3 Criteria for Diagnosis of IPF

ILD, interstitial lung disease; CTD, connective tissue disease; UIP, usual interstitial pneumonia; HRCT, high-resolution computed tomography.
TABLE 17-4 Summary of Features of the Idiopathic Interstitial Pneumonias
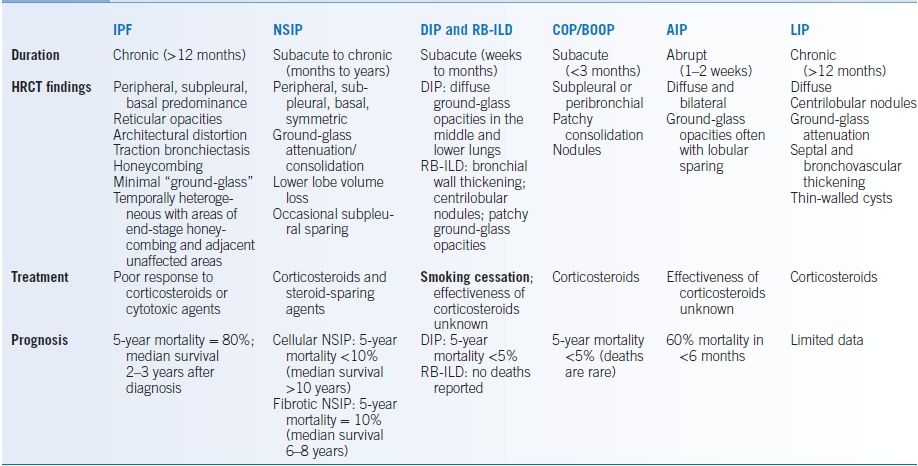
AIP, acute interstitial pneumonia; BOOP, bronchiolitis organizing pneumonia; COP, cryptogenic organizing pneumonia; DIP, desquamative interstitial pneumonia; HRCT, high-resolution computed tomography; IPF, idiopathic pulmonary fibrosis; LIP, lymphocytic interstitial pneumonia; NSIP, nonspecific interstitial pneumonitis; RB-ILD, respiratory bronchiolitis-interstitial lung disease.
Data from King TE. Clinical advances in the diagnosis and therapy of the interstitial lung diseases. Am J Respir Crit Care Med 2005;172:268–279.
Differential Diagnosis
- IPF is histologically identical to UIP, but with no known inciting agent. It typically affects patients >50 years of age and men much more commonly than women. Diagnosis of IPF can be made without a surgical lung biopsy (Table 17-3).6 Distinguishing IPF from fibrosing NSIP may be very difficult.
- NSIP is a pattern of lung response to a variety of injuries. Diseases associated with NSIP include CTD such as scleroderma, rheumatoid arthritis, and polymyositis-dermatomyositis. Presentation is similar to IPF although NSIP may be a more subacute presentation, and patients may have signs and symptoms of a CTD. CXR is similar to other idiopathic interstitial pneumonias, and HRCT characteristically shows ground-glass opacities with varying degrees of fibrosis. There are three histologic subgroups: cellular (Group I), mixed cellular and fibrosis (Group II), and fibrosis (Group III).
- COP is defined histologically by BOOP, which encompasses collagenous granulation tissue in the lumens of small airways and alveolar ducts with surrounding alveolar chronic inflammation. It classically presents like pneumonia, but the patient fails to respond to antibiotics. BOOP may be caused by drugs, inhalational exposures, or infection. COP can be diagnosed only if known causes of BOOP are excluded. CXR and HRCT show patchy peripheral infiltrates that may be migratory. The diagnosis can usually be made by transbronchial biopsy.
- Sarcoidosis is the second most common ILD. Infectious granulomatous disease should be ruled out before initiating therapy. HRCT typically reveals nodular infiltrates in a lymphatic distribution with an upper and midlung predominance. Mediastinal and bilateral hilar lymphadenopathy is common.
- HP is also known as extrinsic allergic alveolitis. Inflammation is caused by repeated inhalation of an inciting agent in a sensitized host. Presentation can be acute, subacute, or chronic depending on the type and level of exposure to the offending agent. Common forms of HP include bird-fancier’s disease, farmer’s lung disease, and hot tub lung, but dozens of others have been reported. HRCT is variable and includes centrilobular nodules, ground-glass opacities, or extensive honeycombing.
- GPA is a necrotizing granulomatous vasculitis of the upper and lower respiratory tract and can cause a rapidly progressive glomerulonephritis. Presentations include sinusitis, epistaxis, ulcers, hemoptysis, and dyspnea. Specificity of cytoplasmic antineutrophilic cytoplasmic antibody (cANCA) testing is 99% but requires confirmation with positive antibody to proteinase 3. Sensitivity ranges from 30% to 60%. There is a strong correlation between cANCA and disease activity.4 Radiographic features are variable but include heterogeneous pulmonary infiltrates and pulmonary nodules. Pathologic specimens include the presence of inflammatory masses with necrotic areas and granulomatous vasculitis.
- There are a variety of eosinophilic lung diseases including acute and chronic eosinophilic pneumonia (AEP and CEP, respectively), allergic bronchopulmonary aspergillosis, Churg-Strauss syndrome, and idiopathic hypereosinophilic syndrome. AEP typically has an acute, severe presentation characterized by diffuse infiltrates. CEP is a slow, progressive disease that may have severe pulmonary and systemic symptoms. Radiographic findings commonly show peripheral pulmonary infiltrates that can cross fissures. Bronchoalveolar lavage (BAL) eosinophils should be >25%, and peripheral eosinophilia is typical.
- PAP is characterized by the accumulation of periodic acid-Schiff–positive, lipid-rich proteinaceous material in the alveolar spaces. Granulocyte macrophage colony–stimulating factor (GM-CSF) mutations or acquired inactivating antibodies play a critical role in the pathogenesis. Disease onset is typically gradual. CXR shows a diffuse alveolar filling process. HRCT shows diffuse ground-glass opacities in a pattern referred to as crazy paving. BAL demonstrates thick milky effluent due to a large amount of proteinaceous material.
- LAM is a rare cystic disease, which causes progressive airflow obstruction in young women. Pneumothoraces are common. HRCT shows numerous uniform thin-walled cysts.
- PLCH is a disease that features activation and proliferation of Langerhans cells. Greater than 90% of cases occur in smokers.4 Symptoms of cough and dyspnea typically occur insidiously. CXR shows diffuse micronodular infiltrates. HRCT shows numerous irregular cysts and centrilobular nodules.
TREATMENT
- The most recent international guidelines do not find evidence to support any pharmacologic therapy for IPF, including corticosteroid monotherapy.6 A recent trial noted increased mortality and morbidity with combination prednisone, azathioprine, and N-acetylcysteine.7 The role of alternative pharmacologic agents, such as acetylcysteine (as monotherapy) or pirfenidone, has yet to be fully elucidated.6 Early referral for lung transplantation is advised, if no contraindications exist.
- NSIP, especially when exhibiting predominately cellular pathology, responds to immunosuppressive therapy and has a significantly better prognosis than IPF.8
- COP tends to be steroid responsive but has a high rate of relapse (up to 50%).4
- First-line treatment for sarcoidosis is steroids, typically 0.5 mg/kg/day, but when and in whom to initiate therapy is still controversial. Generally, steroids for chronic ILD are not initiated unless significant pulmonary symptoms exist and fail to remit after an extended period of time (e.g., 6 to 12 months). Extrapulmonary disease (ophthalmologic, cardiac, and CNS) usually requires treatment. Steroid-sparing agents such as methotrexate, imuran, and leflunomide can be considered in patients unable to wean from high-dose steroids. Tumor necrosis factor-alpha (TNF-α) inhibitors can be considered in refractory disease.9
- HP treatment includes removal of the offending exposure. Steroids can be used if very symptomatic.
- The mainstay of treatment for GPA was oral cyclophosphamide plus corticosteroids; however, recent evidence suggests that rituximab with corticosteroids is as effective at inducing remission in severe disease. Untreated disease follows a rapidly fatal course.4,10
- AEP often responds to corticosteroids. CEP is also responsive to corticosteroids but may relapse in approximately 50% of patients.
- Spontaneous recovery from PAP occurs in >25% of patients. Corticosteroids have no clear benefit.4 Subcutaneous GM-CSF has shown some benefit. In patients with severe respiratory failure, repetitive whole-lung lavage with saline is necessary.
- LAM treatment options include antiestrogen agents, oophorectomy, and sirolimus.11
- Smoking cessation is beneficial for PLCH. Corticosteroid therapy early in the disease may be helpful.
Pulmonary Hypertension
GENERAL PRINCIPLES
- Pulmonary hypertension (PH) is frequently encountered during the evaluation of respiratory or cardiac conditions.
- While mild PH (i.e., PASP <50 or mean PAP <30) can occur in the setting of acute conditions (e.g., pulmonary embolism, pulmonary edema, acute respiratory distress syndrome), chronic PH should prompt an evaluation to determine its origins and guide therapy.
- Internists and primary care physicians should be aware of basic pulmonary physiology, the differential diagnosis of PH, and how to evaluate the condition in order to identify which patients need therapeutic intervention.
- PH is defined as mean PAP ≥25 mm Hg at rest.
- There are five main groups of PH delineated in the 2008 Dana Point clinical classification scheme (Table 17-5).12,13 Mechanisms leading to PH differ among categories:
- Group I encompasses conditions distinguished by severe vascular remodeling.
- Group II patients develop pulmonary venous hypertension from elevated downstream pressures in the left side of the heart.
- Group III patients encounter lung destruction as a result of underlying lung disease and/or vascular remodeling due to chronic hypoxemia.
- Group IV disease results from progressive obliteration of vasculature due to embolization of foreign material.
- Group V conditions have variable mechanisms for developing PH, including vasculature compression, lung destruction, or vascular remodeling.
- Group I encompasses conditions distinguished by severe vascular remodeling.
- Pulmonary arterial hypertension (PAH) is a category of diseases that share pathobiology resulting from vasoconstriction, endothelial and smooth cell proliferation, and in situ thrombosis.
- Prevalence of PAH is estimated to be 15 to 26 cases per million adults.14,15
- PAH is defined as elevated pulmonary artery pressures in the setting of normal LV filling pressures (i.e., PAOP ≤15 mm Hg).
- Notable for severely elevated pulmonary artery pressures that can ultimately result in right ventricular failure.
- Most common types are idiopathic PAH and PAH associated with collagen vascular diseases, particularly progressive systemic sclerosis or scleroderma.
- Prevalence of PAH is estimated to be 15 to 26 cases per million adults.14,15
- Pulmonary venous hypertension is the most frequently encountered type of PH in western countries, while PH associated with lung disease and/or hypoxemia is second most common.
- The leading cause of death in patients with PAH is right heart failure.
TABLE 17-5 2008 Dana Point Clinical Classification of Pulmonary Hypertension (PH)

BMPR-2, bone morphogenic protein receptor, type 2; ALK-1, activin receptor-like kinase 1 gene; PLCH, pulmonary Langerhans cell histiocytosis; LAM, lymphangioleiomyomatosis.
Data from Galie N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009;30:2493–2537.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


