FIGURE 20-1. Distribution of body water in a term newborn infant. (Reproduced, with permission, from Bell EF, Oh W. Fluid and electrolyte management. In: MacDonald MG, Seshia MMK, Mullett MD, eds. Avery’s Neonatology: Pathophysiology and Management of the Newborn. 6th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005:363.)
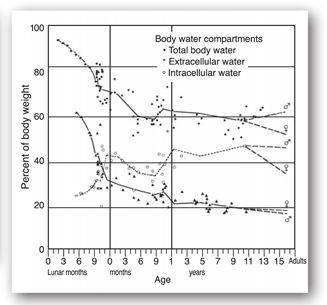
FIGURE 20-2. Changes in body water from early fetal life to adult life. (Reproduced, with permission, from Friis-Hansen B. Water distribution in the foetus and newborn infant. Acta Paediatr Scand. 1983;305:8.)
The electrolyte composition of ECW versus ICW is very different (Figure 20-3). Sodium is the major cation found in intravascular water (plasma volume) of the ECW. Potassium, calcium, and magnesium make up a much smaller amount of the intravascular cations. Chloride is the primary intravascular anion and bicarbonate, protein, and other anions comprise the balance. The electrolyte composition of the interstitial component of ECW is similar to the intravascular composition, but protein content is lower. Potassium and magnesium are the major cations found in ICW. Phosphate (organic and inorganic) is the primary intracellular anion, and bicarbonate makes up a smaller amount.18
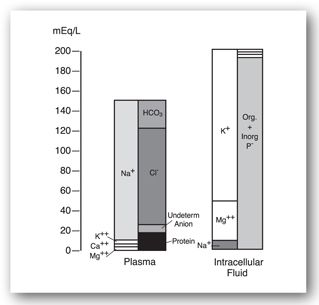
FIGURE 20-3. Ion distribution in the blood plasma, which represents extracellular fluid, and in the intracellular fluid compartment. (Reproduced, with permission, from Bell EF, Oh W. Fluid and electrolyte management. In: MacDonald MG, Seshia MMK, Mullett MD, eds. Avery’s Neonatology: Pathophysiology and Management of the Newborn. 6th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005:364.)
These compositional differences in ECW and ICW, along with the age-related differences in the amounts of these water compartments, can result in maturational differences in the amount of electrolytes per kg of body weight. For example, since premature neonates have a larger ECW compartment and ECW contains a higher amount of sodium and chloride, premature neonates contain a higher amount of sodium and chloride per kilogram of body weight compared to term neonates.18 These principles are important to keep in mind when managing neonatal fluid and electrolyte therapy. One must also remember that the management of fluid and electrolyte therapy in the mother during labor can result in alterations in the newborn’s fluid and electrolyte status. For example, if the mother is given too much fluid (i.e., too much free water) during labor, the newborn may be born with hyponatremia.21
Insensible water loss is the water that is lost via evaporation from the skin and through the respiratory tract.18 Knowledge of the factors that influence insensible water loss in pediatric patients is important to estimate appropriate water intake and to assess electrolyte imbalances that may occur. Compared to adults, neonates and young infants have an increase in the amount of insensible water loss. This is primarily due to their increased surface area to body weight ratio and higher respiratory rate. Smaller newborns and those born at a younger gestational age (GA) have an even higher insensible water loss. This is related to their immature (thinner) skin, greater skin blood flow, and larger TBW. Many other factors increase insensible water loss, such as the environmental and body temperature, radiant warmers, phototherapy, motor activity, crying, and skin breakdown or injury. Congenital skin defects, such as gastroschisis, omphalocele, or neural tube defects will also increase insensible water loss. The use of high inspired or ambient humidity, plastic heat shields or blankets, occlusive dressings, and topical waterproof agents will decrease insensible water loss.
The primary functions of the kidney (glomerular filtration, tubular secretion, and tubular reabsorption) are all decreased in the newborn, especially in the premature newborn, compared to adults. These functions increase with GA at birth and with postnatal age (PNA). The decreased glomerular and tubular functions in the neonatal kidney result in differences in how the neonate handles various electrolyte loads and differences in the normal reference ranges for several electrolytes, as described below.
Sodium
Normal range22 for premature neonates (at 48 hr of life): 128–148 mEq/L or 128–148 mmol/L
newborns: 133–146 mEq/L or 133–146 mmol/L
infants: 139–146 mEq/L or 139–146 mmol/L
children: 138–145 mEq/L or 138–145 mmol/L
adults: 136–142 mEq/L or 136–142 mmol/L
Sodium is primarily excreted via the kidneys, but it is also excreted via stool and sweat.23 Usually, unless diarrhea occurs, sodium loss in the stool is minimal. In children with cystic fibrosis, aldosterone deficiency, or pseudohypoaldosteronism, the sodium concentration in sweat is increased and higher sweat losses may contribute to or cause sodium depletion.
In neonates and young infants, the renal handling of sodium is altered compared to adults.24,25 Differences in tubular reabsorption, aldosterone concentrations, and patterns of renal blood flow help to maintain a positive sodium balance, which is required for growth. In the neonate, sodium reabsorption is decreased in the proximal tubule, but increased in the distal tubule. Aldosterone increases sodium reabsorption in the distal tubules, and plasma concentrations of renin, angiotensin II, and aldosterone are all increased in neonates. This increase in aldosterone may be a compensatory mechanism to help increase sodium reabsorption in the distal tubule. The pattern of renal blood flow is also different in the neonate. In adults, a larger amount of renal blood flow goes to the cortical area of the kidneys. However, in the neonate, the majority of renal blood flow goes to the medullary area, which is more involved with sodium conservation than excretion. These factors help the neonatal kidney to retain sodium, but also result in the neonate having a decreased ability to excrete a sodium load. Therefore, if an excessive amount of sodium is administered to a neonate, it will result in sodium retention with subsequent water retention and edema.
Although most infants are in a positive sodium balance, very low birth weight infants (birth weight <1.5 kg) are usually in a negative sodium balance.24 This is due to their very immature kidneys and the larger amounts of sodium that are lost in the urine. These infants are at a higher risk of sodium imbalance and may require higher amounts of sodium, especially during the first weeks of life.
Compared to adults, pediatric patients may be more susceptible to imbalances of sodium and water. This may be due to their higher amount of TBW and the common pediatric occurrence of causative factors such as diarrhea and dehydration.
Hyponatremia
In infants and children, hyponatremia is defined as a serum sodium less than 135 mEq/L, although slightly lower values would be considered acceptable for premature neonates and newborns.23 As in adults, hyponatremia occurs in pediatric patients when the ratio of water to sodium is increased. This may occur with low, normal, or high amounts of sodium in the body; likewise, the amount of water in the body may be low (hypovolemic), normal (euvolemic), or high (hypervolemic). The causes of hyponatremia in pediatric patients are the same as in adults. However, certain causes may be more commonly seen in children.
In hypovolemic hyponatremia, both sodium and water have been lost from the body, but a higher proportion of sodium has been lost. The most common cause of hypovolemic hyponatremia in children is diarrhea due to gastroenteritis.23 Emesis can also cause hyponatremia if hypotonic fluids are administered, but most children with emesis have either a normal serum sodium or hypernatremia. In addition to gastrointestinal (GI) losses, hypovolemic hyponatremia may also occur from losses of sodium through the skin (e.g., excessive sweating or burns), third space losses, and renal losses.
Renal sodium loss can occur in the pediatric population from a number of causes including thiazide or loop diuretics, osmotic diuresis, cerebral salt wasting, and hereditary or acquired kidney diseases. Cerebral salt wasting is thought to be due to hypersecretion of atrial natriuretic peptide, which causes renal salt wasting. This condition is usually seen in patients with central nervous system disorders such as head trauma, brain tumors, hydrocephalus, cerebral vascular accidents, neurosurgery and brain death.26 Hereditary kidney diseases that can cause hypovolemic hyponatremia include juvenile nephronophthisis, autosomal recessive polycystic kidney disease, proximal (type II) renal tubular acidosis, 21-hydroxylase deficiency, and pseudohypoaldosteronism type I. Patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency have an absence of aldosterone. Aldosterone is needed for sodium retention and potassium and acid excretion in the kidneys. The lack of aldosterone in these patients produces hyponatremia, hyperkalemia, and metabolic acidosis. Patients with pseudohypoaldosteronism have elevated aldosterone serum concentrations, but the kidneys do not respond properly to aldosterone. A lack of response to aldosterone by the renal tubules may also occur in children with a urinary tract obstruction and/or acute urinary tract infection and result in hyponatremia.23
In euvolemic hyponatremia, patients have no real evidence of volume depletion or volume overload.23 Usually, these patients have a slight decrease in total body sodium with an excess of TBW. Although some patients may have an increase in body weight (indicating volume overload), patients often appear clinically normal or have subtle signs of fluid overload. Causes of euvolemic hyponatremia include the syndrome of inappropriate antidiuretic hormone (SIADH), glucocorticoid deficiency, hypothyroidism, and water intoxication. Although SIADH is not common in children, it may occur in patients with central nervous system disorders or lung disease and tumors. Certain medications can cause an increase in antidiuretic hormone (ADH) secretion and are reviewed in Chapter 6: Electrolytes, Other Minerals, and Trace Elements.
Dilutional hyponatremia may commonly occur in hospitalized children who receive relatively large amounts of free water (e.g., hypotonic IV solutions). This may even occur when medications are diluted in 5% dextrose in water, for example, and administered as 50- or 100-mL IV rider bags or piggyback riders. Neonates and young infants are more prone to this water overload (due to their lower glomerular filtration rate [GFR] and limited ability to excrete water), and, thus, should receive medications diluted in smaller volumes of IV fluid. Other causes of hyponatremia due to water intoxication in pediatric patients include administration of diluted infant formula, tap water enemas, infant swimming lessons, forced water intake (child abuse), and psychogenic polydipsia.23 (See Minicase 1.)
A Case of Hyponatremia and Seizures
HUNTER N., A 3-DAY-OLD MALE, is currently in the neonatal ICU being treated with antibiotics for suspected sepsis. This morning Hunter. N. began having rhythmic clonic twitching of his lower extremities, fluttering of his eyelids, and repetitive chewing movements, consistent with seizure activity. Hunter N. was born at 39 weeks of gestation to a mother with prolonged rupture of membranes (>72 hours). On the day of his birth, Hunter N. was admitted to the neonatal ICU with an elevated temperature, tachycardia (HR 166), and low WBC count (3.2 × 103 cells/mm3). Blood and urine cultures were obtained and antibiotics were started to treat his possible sepsis. Culture results are still pending. Medications include ampicillin 85 mg IV in 25 mL D5W as IV rider q 8 hr (75 mg/kg/day) and gentamicin 8.5 mg IV in 25 mL D5W as IV rider q 12 hr (5 mg/kg/day).
Hunter N.’s vital signs include BP 76/46 mm Hg, HR 129 beats/min, RR 35 breaths/min, and temperature 98.8°F. Length is 49 cm (50th percentile for age), and weight is 3.4 kg (50th percentile for age). Laboratory data includes sodium 119 mEq/L, potassium 3.8 mEq/L, chloride 99 mEq/L, total CO2 20 mEq/L, BUN 9 mg/dL, SCr 0.7 mg/dL, and glucose 87 mg/dL.
Question: What is the most likely cause of Hunter N.’s seizure activity and electrolyte imbalance? What other laboratory tests should be obtained to further assess his seizure disorder?
Discussion: Electrolyte imbalance is a common cause of neonatal seizures. As in adults, hyponatremia may cause seizure activity in neonates and occurs when the ratio of water to sodium is increased. The total body content of sodium in patients with hyponatremia may be low, normal, or high, and the volume status may be hypovolemic, euvolemic, or hypervolemic. There are many causes of hyponatremia, but the most likely cause in Hunter N. is the extra D5W that he received with his antibiotics. Dilutional hyponatremia may occur in neonates and young infants when medications are administered in excess fluids, such as IV riders of 5% dextrose in water. These patients are more prone to water overload due to their lower GFR and their limited ability to excrete water. Medications for these patients should be diluted in smaller amounts of IV fluid, so that excess fluid is not administered. To better define Hunter N.’s sodium and volume status, his total fluid intake and output, type of IV fluids administered, changes in body weight, and other laboratory data need to be assessed. In addition, other causes of hyponatremia, such as meningitis and SIADH, should be ruled out. It should be noted that a low WBC count, as observed in Hunter N., often occurs in neonates with a serious bacterial infection (see section on White Blood Cell Count). Although the most likely cause of Hunter N.’s seizure activity is his low serum sodium, his serum calcium, phosphorous, and magnesium levels should also be assessed, as other electrolyte abnormalities can also cause seizure activity.
In hypervolemic hyponatremia, both sodium and water are increased in the body, but there is a greater increase in water than sodium. Hypervolemic hyponatremia is typically observed in patients with congestive heart failure (CHF), cirrhosis, nephrotic syndrome, and chronic renal failure. It may also occur in patients with hypoalbuminemia or in patients with capillary leak syndrome due to sepsis.23 These conditions decrease the patient’s effective blood volume, either due to poor cardiac function or third spacing of fluid. The compensatory mechanisms in the body sense this decrease in blood volume; ADH and aldosterone are secreted and cause retention of water and sodium in the kidneys. A decrease in serum sodium occurs because the intake of water in these patients is greater than their sodium intake and ADH decreases water excretion.
Hypernatremia
In general for pediatric patients, hypernatremia is defined as a serum sodium concentration greater than 145 mEq/L. As in adults, hypernatremia occurs in pediatric patients when the ratio of sodium to water is increased. This may occur with low, normal, or high amounts of sodium in the body. Hypernatremia may occur with excessive sodium intake, excess water loss, or a combination of water and sodium loss when the water loss exceeds the sodium loss.23
Excessive sodium intake or sodium intoxication may occur due to improperly mixed infant formulas, excess sodium bicarbonate administration, IV hypertonic saline solutions, intentional salt poisoning (e.g., child abuse), and ingestion of sodium chloride or seawater.23 Neonates, especially premature newborns, and young infants can develop hypernatremia from excessive sodium due to the decreased ability of immature kidneys to excrete a sodium load. This becomes a problem especially in the premature neonate when IV sodium bicarbonate is used to correct a metabolic acidosis.
Excess water loss resulting in hypernatremia may occur in pediatric patients due to diabetes insipidus, increased insensible water losses, or inadequate intake. Diabetes insipidus can be of central or nephrogenic origin and either type can be acquired or congenital. Also, certain drugs may cause diabetes insipidus (see Chapter 6: Electrolytes, Other Minerals, and Trace Elements).
Neonates may be predisposed to hypernatremia from increased insensible water losses, especially during the first few days of life. A normal physiologic contraction of the ECW occurs after birth, resulting in a net loss of water and sodium. In term infants, this may result in a weight loss of 5% to 10% during the first week of life. In premature newborns, the weight loss may be 10% to 20%. This water loss, plus the relatively large and variable insensible water loss in neonates, can complicate the assessment of fluid and sodium balance. More premature newborns may be at higher risk for hypernatremia, as they have a more pronounced contraction of ECW and higher insensible water loss.27 The use of radiant warmers and phototherapy (used to treat hyperbilirubinemia) will further increase insensible water loss.
Inadequate water intake can also cause hypernatremia in pediatric patients. This may be due to the caregiver not administering enough fluids (e.g., child neglect or abuse, or ineffective breast-feeding). Ineffective breast-feeding may result in severe hypernatremic dehydration. Rarely, inadequate intake may be due to adipsia (absence of thirst).23
Hypernatremia, due to water losses greater than sodium losses, occurs in patients with water and sodium losses through the GI tract (e.g., diarrhea, emesis, nasogastric suctioning, and osmotic cathartics), skin (e.g., burns and excessive sweating), and kidneys (e.g., diabetes mellitus, chronic kidney disease, osmotic diuretics, and acute tubular necrosis [polyuric phase]). Hypernatremia is most likely to occur in infants or children with diarrhea who also have inadequate fluid intake due to anorexia, emesis, or lack of access to water.
It should be noted that due to the immaturity of the blood vessels in their central nervous system, premature neonates are especially vulnerable to the adverse effects of hypernatremia (e.g., intracranial hemorrhage). These patients are also at greater risk of adverse central nervous system effects if an elevated serum sodium is corrected too rapidly. Thus, maintaining a proper sodium balance in these patients is extremely important.
Potassium
Normal range11,22 for premature neonates (at 48 hr of life): 3.0–6.0 mEq/L or 3.0–6.0 mmol/L
newborns: 3.7–5.9 mEq/L or 3.7–5.9 mmol/L
infants: 4.1–5.3 mEq/L or 4.1–5.3 mmol/L
children: 3.4–4.7 mEq/L or 3.4–4.7 mmol/L
adults: 3.8–5.0 mEq/L or 3.8–5.0 mmol/L
Potassium is the major intracellular cation, and less then 1% of total body potassium is found in the plasma.23 However, small changes in serum potassium can have large effects on cardiac, neuromuscular, and neural function. Thus, appropriate homeostasis of extracellular potassium is extremely important. Insulin, aldosterone, acid–base balance, catecholamines, and renal function all play important roles in the regulation of serum potassium. Serum potassium can be lowered quickly when potassium shifts intracellularly or more slowly via elimination by the kidneys.
The kidney is the primary organ that regulates potassium balance and elimination. Potassium undergoes glomerular filtration and almost all filtered potassium is then reabsorbed in the proximal tubule. Urinary excretion of potassium, therefore, is dependent on distal potassium secretion by the collecting tubules. Neonates and young infants, however, have a decreased ability to secrete potassium via the collecting tubules. Thus, the immature kidneys tend to retain potassium. This results in a positive potassium balance, which is required for growth (potassium is incorporated intracellularly into new tissues).24,25 Potassium retention by the immature kidneys also results in higher serum potassium concentrations compared to the adult.25
Hypokalemia
Hypokalemia is defined as a serum potassium concentration <3.5 mEq/mL. As in adults, a low serum potassium may occur in pediatric patients due to an intracellular shift of potassium, decreased intake, or increased output (from renal or extrarenal losses). An intracellular shift of potassium may be seen with alkalosis, beta-adrenergic stimulation, or insulin treatment. Endogenous beta-adrenergic agonists (such as epinephrine released during stress) and exogenously administered beta-agonists (such as albuterol) stimulate the cellular uptake of potassium. Other causes of an intracellular shift of potassium seen in pediatric patients include overdoses of theophylline, barium intoxication, and glue sniffing (toluene intoxication). A falsely low potassium concentration can be reported in a patient with a very elevated white blood cell (WBC) count (e.g., a patient with leukemia) if the plasma sample is inappropriately stored at room temperature. This allows the WBCs to uptake potassium from the plasma resulting in a falsely low measurement.23
Most cases of hypokalemia in children are related to extrarenal losses of potassium due to gastroenteritis and diarrhea.23 Hypokalemia due to diarrhea is usually associated with a metabolic acidosis, since bicarbonate is also lost in the stool. Adolescent patients with eating disorders may be hypokalemic due to inadequate intake of potassium, for example, in patients with anorexia nervosa. Adolescents with bulimia or laxative abuse may also have significant extrarenal losses of potassium.
Many causes of hypokalemia due to renal potassium loss exist. Medications commonly used in the pediatric population that are associated with hypokalemia due to renal potassium loss include loop and thiazide diuretics, corticosteroids, amphotericin B, and cisplatin (see Chapter 6: Electrolytes, Other Minerals, and Trace Elements). Cushing syndrome, hyperaldosteronism, and licorice ingestion may also cause hypokalemia via this mechanism.
In the pediatric population, other causes of increased renal potassium loss, such as hereditary diseases, must be considered. Remember that many hereditary diseases are first diagnosed during infancy and childhood. Renal tubular acidosis (both distal and proximal types) may present with hypokalemia and metabolic acidosis. Patients with cystic fibrosis have greater losses of chloride in sweat. This may lead to metabolic alkalosis, low urine chloride, and hypokalemia. Certain forms of congenital adrenal hyperplasia may also lead to increased renal potassium excretion and hypokalemia. Other inherited renal diseases that are due to defects in renal tubular transporters, such as Bartter syndrome, may result in metabolic alkalosis, hypokalemia, and high urine chloride. Thus, unlike the adult population, hereditary diseases need to be considered when certain electrolyte abnormalities are not explained by common causes.
Hyperkalemia
In infants, children, and adults, hyperkalemia is defined as a serum potassium greater than 5.0 mEq/L. Since a normal serum potassium is slightly higher in neonates and preterm infants, hyperkalemia is defined as a serum potassium greater than 6.0 mEq/L in these patients. Hyperkalemia is one of the most alarming electrolyte imbalances because it has the potential to cause lethal cardiac arrhythmias.
As in adults, hyperkalemia in pediatric patients may be due to increased intake, an extracellular shift of potassium, or decreased renal excretion. Factitious hyperkalemia is very common in pediatric patients, due to the difficulty in obtaining blood samples. Hemolysis often occurs during blood sampling and potassium is released from red blood cells (RBCs) in sufficient amounts to cause falsely elevated test results. This may especially happen with improperly performed heelsticks (see section on Pediatric Blood Sampling). Potassium may also be released locally from muscles after prolonged tourniquet application or from fist clenching, which may also result in false elevations of measured potassium. A falsely elevated serum potassium can also be observed in patients with leukemia or extremely elevated WBC counts (usually >200,000/mm3) due to the release of potassium from WBCs. Prompt analysis with measurement of a plasma sample usually avoids this problem.23
Hyperkalemia may occur due to extracellular shifts of potassium. During a metabolic acidosis, hydrogen ions move into the cells (down a concentration gradient), and in exchange, potassium ions move out of the cells into the extracellular (intravascular) space. This shift leads to a significant increase in serum potassium.
In older patients with fully developed (normal) renal function, hyperkalemia rarely results from increased intake alone. However, this may occur in patients receiving large amounts of oral or IV potassium or in patients receiving rapid or frequent blood transfusions (due to the potassium content of blood).23 In patients with immature renal function or in those with renal failure, increased intake of potassium can also lead to hyperkalemia due to decreased potassium excretion.
Decreased renal excretion of potassium is the most common cause of hyperkalemia. Decreased potassium excretion occurs in patients with immature renal function, renal failure, primary adrenal disease, hyporeninemic hypoaldosteronism, renal tubular disease, and with certain medications.23 Hyperkalemia is the most common life-threatening electrolyte imbalance seen in neonates. Due to the decreased ability of immature kidneys to excrete potassium, neonates, particularly premature neonates, may be predisposed to hyperkalemia. These patients also cannot tolerate receiving extra potassium. Hyperkalemia can be seen in premature infants, during the first 3 days of life, even when exogenous potassium is not given and when renal dysfunction is absent.27 A rapid elevation in serum potassium is seen within the first day of life in more immature newborns. This hyperkalemia, which can be life-threatening, may be due to a shift of potassium from the intracellular space to the extracellular (intravascular) space, immaturity of the distal renal tubules, and a relative hypoaldosteronism.28
Acute or chronic renal failure in pediatric patients will decrease potassium excretion and may result in hyperkalemia. Several inherited disorders may also cause decreased potassium excretion and hyperkalemia in pediatric patients including certain types of congenital adrenal hyperplasia (e.g., 21-hydroxylase deficiency), aldosterone synthase deficiency, sickle cell disease, and pseudohypoaldosteronism (types I and II).23 Medications used in pediatric patients that may also cause hyperkalemia include angiotensin-converting enzyme inhibitors, beta2-adrenergic antagonists, potassium-sparing diuretics, nonsteroidal anti-inflammatory agents, heparin, trimethoprim, and cyclosporine.
Serum Bicarbonate (Total Carbon Dioxide)
Normal range22,25 for
preterm infants: 16–20 mEq/L or 16–20 mmol/L
full-term infants: 19–21 mEq/L or 19–21 mmol/L
infants–children 2 yr of age: 18–28 mEq/L or 18–28 mmol/L
children >2 yr and adults: 21–28 mEq/L or 21–28 mmol/L
The total carbon dioxide concentration actually represents serum bicarbonate, the basic form of the carbonic acid–bicarbonate buffer system (i.e., a low serum bicarbonate may indicate an acidosis). In addition to the buffer systems, the kidneys also play an important role in acid–base balance. The proximal tubule reabsorbs 85% to 90% of filtered bicarbonate. The distal tubule is responsible for the net secretion of hydrogen ions and urinary acidification.29 Compared to adults, neonates have a decreased capacity to reabsorb bicarbonate in the proximal tubule, and, therefore, a decreased renal threshold for bicarbonate (the renal threshold is the serum concentration at which bicarbonate appears in the urine). The mean renal threshold for bicarbonate in adults is 24–26 mEq/L but only 18 mEq/L in the premature infant and 21 mEq/L in the term neonate. The renal threshold for bicarbonate increases during the first year of life and reaches adult values by about 1 year of age. Neonates also have decreased function of the distal tubules to secrete hydrogen ions and to acidify urine. The ability to acidify urine increases to adult values by about 1–2 months of age.25,29 The neonate’s decreased renal capacity to reabsorb bicarbonate and excrete hydrogen ions results in lower normal values for serum bicarbonate and blood pH. In addition, the neonate is less able to handle an acid load or to compensate for acid–base abnormalities.
It should be noted that for multiple reasons, the full-term newborn is in a state of metabolic acidosis immediately after birth (arterial pH 7.11–7.36). The blood pH increases to more normal values within 24 hours, mostly due to increased excretion of carbon dioxide via the lungs.25
Calcium
Total serum calcium—normal range12 for newborns:
3–24 hr old: 9.0–10.6 mg/dL or 2.3–2.65 mmol/L
24–48 hr old: 7.0–12.0 mg/dL or 1.75–3.0 mmol/L
4–7 days old: 9.0–10.9 mg/dL or 2.25–2.73 mmol/L
children: 8.8–10.8 mg/dL or 2.2–2.7 mmol/L
adolescents and adults: 8.4–10.2 mg/dL or 2.1–2.55 mmol/L
Ionized calcium—normal range for newborns:
3–24 hr old: 4.3–5.1 mg/dL or 1.07–1.27 mmol/L
24–48 hr old: 4.0–4.7 mg/dL or 1.00–1.17 mmol/L
infants, children, adolescents, and adults: 4.5–4.92 mg/dL or 1.12–1.23 mmol/L
Calcium plays an integral role in many physiologic functions including muscle contraction, neuromuscular transmission, blood coagulation, bone metabolism, and regulation of endocrine functions. The great majority of calcium in the body (99%) is found in the bone, primarily as hydroxyapatite. Due to the growth that occurs during infancy and childhood, bone mass increases faster than body weight.30 This increase in bone mass requires a significant increase in total body calcium. The increased calcium requirement is reflected in the higher recommended daily allowances (per kg body weight) in pediatric patients compared to adults.
Calcium regulation in the body has two main goals.30 First, serum calcium must be tightly regulated to permit the normal physiologic functions in which calcium plays a role. Second, calcium intake must be adequate to permit appropriate bone mineralization and skeletal growth. It is important to remember that bone mineralization may be sacrificed (i.e., calcium may be released from the bone) in order to allow maintenance of a normal serum calcium concentration.
As in adults, serum calcium in pediatric patients is regulated by a complex hormonal system that involves vitamin D, serum phosphate, parathyroid hormone (PTH), and calcitonin. Briefly, calcium is absorbed in the GI tract, primarily via the duodenum and jejunum.30 Although some passive calcium absorption occurs when dietary intake is high, most GI absorption of calcium occurs via active transport that is stimulated by 1,25-dihydroxyvitamin D. This occurs especially when dietary intake is low. Calcium excretion is controlled by the kidneys and influenced by multiple hormonal mediators (e.g., PTH, 1,25-dihydroxyvitamin D, and calcitonin). In the mature kidneys, approximately 99% of filtered calcium is reabsorbed by the tubules with the majority (>50%) absorbed by the proximal tubules. Calcium is also absorbed in the loop of Henle, distal tubule, and collecting ducts.
During the first week of life, urinary calcium excretion is inversely related to GA (i.e., more premature infants will have a greater urinary calcium excretion).25 Compared to adults, urinary calcium excretion is higher in neonates and preterm infants. The urinary calcium-to-creatinine ratio is 0.11 in adults but may be greater than 2 in premature neonates and ranges from 0.05–1.2 in full-term neonates during the first week of life. This high rate of calcium excretion may be related to the immaturity of the renal tubules and may contribute (along with other factors) to neonatal hypocalcemia. In addition, certain medications that are commonly administered to neonates and premature infants, such as furosemide, dexamethasone, and methylxanthines, further increase urinary calcium excretion. These medications may also increase the risk for hypocalcemia as well as nephrocalcinosis and nephrolithiasis.25
Measurement of Calcium
Total serum calcium measures all three forms of extracellular calcium: complex bound, protein bound, and ionized. However, ionized calcium is the physiologically active form. Usually a parallel relationship exists between the ionized and total serum calcium concentrations. However, in patients with alterations in acid–base balance or serum proteins, the ionized serum calcium and total serum calcium are affected, respectively, and measurements of total serum calcium may no longer reflect the ionized serum concentration. Neonates have lower serum concentrations of protein (including albumin) and may be acidotic. This results in a lower total serum calcium concentration for a given ionized plasma concentration.27 Although equations exist to adjust total serum calcium measurements for low concentrations of serum albumin, these equations have limitations and may not be precise. Therefore, ionized calcium should be measured in neonates (if micro-techniques are available) and other pediatric patients with hypoalbuminemia or acid–base disorders.
Hypocalcemia
As in adults, hypocalcemia may occur in pediatric patients due to a variety of causes including inadequate calcium intake, hypoparathyroidism, vitamin D deficiency, renal failure, redistribution of plasma calcium (e.g., hyperphosphatemia and citrated blood transfusions), and hypomagnesemia. Hypocalcemia may also occur due to lack of organ response to PTH (e.g., pseudohypoparathyroidism) and in the neonate due to other specific causes.
In the pediatric population, hypocalcemia most commonly occurs in neonates. Early neonatal hypocalcemia occurs during the first 72 hours of life and may be due to several factors. During fetal development, a transplacental active transport process maintains a higher calcium concentration in the fetus compared to the mother. After birth, this transplacental process suddenly stops. Serum calcium concentrations then decrease, even in healthy full-term newborns, reaching a nadir at 24 hours.30 The high serum calcium concentrations in utero may also suppress the fetus’ parathyroid gland. Thus, early neonatal hypocalcemia may also be due to a relative hypoparathyroidism in the newborn. In addition, newborns may have a decreased response to PTH.
Early neonatal hypocalcemia is more likely to occur in premature and low birth weight newborns. It also occurs more commonly in infants of diabetic mothers, infants with intrauterine growth retardation, and in newborns that have undergone prolonged difficult deliveries. Inadequate calcium intake in critically ill newborns also contributes to hypocalcemia.
Late neonatal hypocalcemia, which usually presents during the first 5–10 days of life, is caused by a high phosphate intake. It is much less common than early neonatal hypocalcemia, especially since the phosphorus content of infant formulas was decreased. It may, however, still occur if neonates are inappropriately given whole cow’s milk. Cow’s milk has a high phosphate load, which can cause hyperphosphatemia and secondary hypocalcemia in the neonate.
Hypocalcemia may also occur in neonates born to mothers with hypercalcemia. The maternal hypercalcemia is usually due to hyperparathyroidism. In utero suppression of the fetal parathyroid gland can lead to hypoparathyroidism and hypocalcemia in the neonate.
Hypocalcemia due to inadequate dietary calcium intake rarely occurs in the United States, but can occur if infant formula or breast milk is replaced with liquids that contain lower amounts of calcium. Hypocalcemia may be iatrogenically induced if inadequate amounts of calcium are administered in hyperalimentation solutions. Adequate amounts of calcium and phosphorus may be difficult to deliver to preterm neonates due to their high daily requirements and limitations of calcium and phosphorus solubility in hyperalimentation solutions. Certain pediatric malabsorption disorders, such as celiac disease, may also cause inadequate absorption of calcium and vitamin D.
Hypoparathyroidism can be caused by many genetically inherited disorders, such as the DiGeorge syndrome, X-linked hypoparathyroidism, or PTH gene mutations.30 These and other syndromes must be considered when pediatric patients present with hypoparathyroidism.
In pediatric patients with vitamin D deficiency, hypocalcemia occurs primarily due to decreased intestinal absorption of calcium. The lower amounts of calcium in the blood stimulate the release of PTH from the parathyroid gland. Parathyroid hormone then prevents significant hypocalcemia via several different mechanisms. It causes bone to release calcium, increases urinary calcium reabsorption, and increases the activity of 1-alpha-hydroxylase in the kidneys (the enzyme that converts 25-hydroxyvitamin D into 1, 25-dihydroxyvitamin D, the active form of vitamin D). Hypocalcemia only develops after these compensatory mechanisms fail. In fact, most children with vitamin D deficiency present with rickets before they develop hypocalcemia.30 In addition to elevated PTH concentrations, children with vitamin D deficiency will have an elevated serum alkaline phosphatase concentration (due to increased osteoclast activity) and a low serum phosphorus (secondary to decreased intestinal absorption and decreased reabsorption in the kidneys), all due to the effects of PTH.
Vitamin D deficiency may be due to several factors including inadequate intake, lack of exposure to sunlight, malabsorption, or increased metabolism of vitamin D (e.g., from medications such as phenobarbital and phenytoin). Generally, patients may have more than one of these factors. For example, institutionalized children (who are not exposed to sunlight) receiving chronic anticonvulsant therapy may be at a greater risk for developing vitamin D deficiency and rickets. Vitamin D deficiency may also occur with liver disease (failure to form 25-hydroxyvitamin D in the liver) and with renal failure (failure to form the active moiety, 1,25-dihydroxyvitamin D, due to a loss of activity of 1-alpha-hydroxylase in the kidneys).
Genetic disorders, such as vitamin D-dependent rickets, may also cause hypocalcemia. The absence of the enzyme, 1-alpha-hydroxylase, in the kidneys occurs in children with vitamin D-dependent rickets type 1. Therefore, these children cannot convert 25-hydroxyvitamin D to its active form. Children with vitamin D-dependent rickets type 2 have a defective vitamin D receptor, which prevents the normal response to 1,25- dihydroxyvitamin D.30 (See Minicase 2.)
Rickets in a Child
RACHEL C., AN 8-YEAR-OLD FEMALE, was admitted to the emergency room from a local pediatric long-term care facility with c/o pain, tenderness, and decreased movement to her right leg. Rachel C. sustained a fall at the long-term care facility when she was being moved from her bed to her wheel chair. Born at term, Rachel C. suffered a traumatic birth with severe perinatal asphyxia. She subsequently developed seizures that were controlled by the combined anticonvulsant therapy of phenobarbital and phenytoin. As a result of her asphyxia at birth, Rachel C. developed spastic cerebral palsy and severe neurodevelopmental delay. She was transferred to the long-term care facility at 6 months of age and has remained on phenobarbital and phenytoin since that time. Two years ago, Rachel C. was diagnosed with gastroesophageal reflux disease (GERD), which has been controlled with antacids. Medications include phenobarbital elixir 40 mg (10 mL) PO BID; phenytoin suspension 50 mg (2 mL) PO TID; and Alternagel® 5 mL PO QID.
Rachel C.’s vital signs include BP 105/69 mm Hg; HR 90/min; RR 22/min; and temperature 98.6°F. Her height is 124 cm (25th percentile for age) and weight is 20 kg (<5th percentile for age). Rachel C.’s physical exam of her chest is significant for a pigeon breast deformity and slightly palpable enlargement of costochondral junctions. She has redness in her right leg, 10 cm below the knee, and pain on movement. The preliminary x-ray findings reveal a fracture of her right tibia with osteomalacia and bone changes consistent with rickets.
Significant laboratory data includes calcium 7.9 mg/dL (normal for children: 8.8–10.8 mg/dL); ionized calcium: 4.0 mg/dL (normal for infants–adults: 4.5–4.92 mg/dL); phosphorus: 2.2 mg/dL (normal for 4–11 years: 3.7–5.6 mg/dL); magnesium: 1.8 mg/dL (normal for 2–14 years: 1.5– 2.3 mg/dL); albumin 2.8 g/dL (normal for children 7–19 years: 3.7–5.6 g/dL); ALT: 58 units/L (normal 10–40 units/L); AST: 68 units/L (normal for children 7–9 years: 15–40 units/L); alkaline phosphatase: 682 units/L (normal for children 2–10 years: 100–320 units/L).
Question: What evidence exists that Rachel C. has rickets? How did her medications affect her serum phosphorus, calcium, and liver enzymes, and how would you modify her drug therapy?
Discussion: Rickets is diagnosed by both radiologic and chemical findings. The preliminary x-ray findings and the physical findings of the pigeon breast deformity (i.e., the sternum and adjacent cartilage appear to be projected forward) and the palpable enlargement of costochondral junctions (rachitic rosary sign) are compatible with the diagnosis of rickets. Serum calcium may be low or normal in patients with rickets, depending on the etiology. The primary causes of rickets in the United States are vitamin D deficiency (with secondary hyperparathyroidism), primary phosphate deficiency, and end-organ resistance to 1,25-dihydroxyvitamin D. In patients with vitamin D deficiency, serum calcium concentrations can be normal or low, phosphorus concentrations are usually low, and alkaline phosphatase activity is elevated. In patients with primary phosphate deficiency, serum calcium is normal, serum phosphorus is low, and alkaline phosphatase is elevated. In patients with end-organ resistance to 1,25-dihydroxyvitamin D, serum calcium is low, serum phosphorus may be low or normal, and serum alkaline phosphatase is elevated.
In Rachel C., ionized calcium and serum phosphorus are both low and serum alkaline phosphatase is high, all of which are consistent with a diagnosis of rickets. Serum magnesium is normal for age; ALT and AST are slightly elevated. The serum magnesium was obtained since hypomagnesemia may also cause hypocalcemia. She also has hypoalbuminemia. A total serum calcium measures all three forms of extracellular calcium: complex bound, protein bound, and ionized. In patients with low albumin, the concentration of ionized calcium will be increased for a given total serum calcium concentration. Equations can be used to “correct” total serum calcium measurements for low concentrations of serum albumin, but these equations have limitations and may not be precise. Thus, in patients with low albumin (like Rachel C.), an ionized serum calcium should be obtained.
Rachel C.’s medications affected her laboratory tests. She is receiving an aluminum-containing antacid, which binds phosphorus in the GI tract. This resulted in decreased absorption of phosphorus and contributed to Rachel C.’s low serum phosphorus. Enzyme-inducing anticonvulsants, such as phenobarbital and phenytoin, will increase the metabolism of vitamin D and may result in a deficiency of vitamin D with resultant anticonvulsant-induced osteomalacia and rickets. Both the aluminum-containing antacid and the anticonvulsants contributed to Rachel C. developing rickets, and thus, to the elevated serum alkaline phosphatase. In addition, due to her other medical conditions, she is nonambulatory and resides at a long-term care facility. Thus, she may have a lack of exposure to sunlight and, therefore, a lack of vitamin D. This lack of vitamin D would also contribute to the development of rickets.
For treatment of her rickets, Rachel C. should be started on oral supplements of calcium, phosphorous, and vitamin D. However, modifications in Rachel C.’s preadmission medications should be made. The aluminum-containing antacid (Alternagel®) should be discontinued and replaced with a calcium-containing antacid (e.g., calcium carbonate). The amount of calcium in this new antacid should then be subtracted from any calcium supplement that would be started in the hospital, so that the total daily dose of calcium stays the same. Alternatively, the total dose of calcium supplement can be given as calcium carbonate. Discontinuing the aluminum-containing antacid will result in a greater amount of phosphorus absorbed enterally. This will then require a decrease in the oral supplement of phosphate (depending on serum phosphorus concentrations). Once Rachel C. is stable, her neurologist should be consulted to see if other anticonvulsants that have less of an enzyme-inducing effect could be used to treat her seizures.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


