FIGURE 5-1. The therapeutic range for a hypothetical drug. Line A is the percentage of patients displaying a therapeutic effect; line B is the percentage of patients displaying toxicity.
Drug concentration monitoring is often criticized by claims that therapeutic ranges are not sufficiently well defined.11,12 The lack of clearly-defined therapeutic ranges for older drugs is partially attributable to how these ranges were originally determined. Eadie describes the process that was typically used for determination of the therapeutic ranges of the antiepileptic drugs: “These ranges do not appear to have been determined by rigorous statistical procedures applied to large patient populations. Rather, workers seem to have set the lower limits for each drug at the concentration at which they perceived a reasonable (though usually unspecified) proportion of patients achieved seizure control, and the upper limit at the concentration above which overdosage-type adverse effects appear to trouble appreciable numbers of patients, the values then being rounded off to provide a pair of numbers, which are reasonably easy to remember.”14 In an ideal world, studies to define therapeutic ranges for drugs should use reliable methods for measurement of response and should be restricted to patients with the same diseases, age range, and concurrent medications.1 In recent years, the Food and Drug Administration (FDA) has recognized the importance of determining concentration versus response relationships early during clinical trials.23
What factors can affect a therapeutic range for a given patient? Anything that affects the pharmacodynamics of a drug, meaning the response at a given drug concentration, will affect the therapeutic range. These factors include
- Indication: Drugs that are used for more than one indication are likely to be interacting with different receptors. Thus, a different concentration versus response profile might be expected depending on the disease being treated. For example, higher serum concentrations of digoxin are needed for treatment of atrial fibrillation as compared to congestive heart failure. Higher antibiotic drug concentrations may be needed for resistant organisms or to penetrate certain infected tissues.
- Active metabolites: As shown in Figure 5-2, variable presence of an active metabolite can shift the therapeutic range for that individual patient up or down. These metabolites may behave in a manner similar to the parent drug or may interact with different receptors altogether. In either case, the relationship between parent drug concentration and response will be altered.
- Concurrent drug treatment: In a manner similar to active metabolites, the presence of other drugs that have similar pharmacodynamic activities will contribute to efficacy or toxicity but not to measurement of the drug concentration. The therapeutic range will be shifted.
- Patient’s age: While there is not much information concerning developmental changes in pharmacodynamics, it is believed that the numbers and affinities of pharmacologic receptors change with progression of age from newborns to advanced age.24 This would be expected to result in a shift of the therapeutic range.
- Electrolyte status: As an example, hypokalemia, hypomagnesemia, and hypercalcemia are all known to increase the cardiac effects of digitalis glycosides and enhance the potential for digoxin toxicity at a given serum concentration.25
- Concurrent disease: As an example, patients with underlying heart disease (cor pulmonale or coronary, atherosclerotic heart disease) have increased sensitivity to digoxin.25 There is also evidence that thyroid disease alters the usual response patterns of digoxin.22
- Variable ratios of enantiomers: Some drugs are administered as racemic mixtures of enantiomers, which may have different response/toxicity profiles as well as pharmacokinetic behaviors. Thus, a given level of the summed enantiomers (using an achiral assay method) will be associated with different levels of response or toxicity in patients with different proportions of the enantiomers. This has been extensively studied for disopyramide.26
- Variable genotype: There is growing evidence that response to certain drugs is genetically determined. For selected drugs, patients may be genotyped before starting drug treatment in order to identify them as nonresponders, responders, or toxic responders (see Chapter 7: Pharmacogenomics and Molecular Testing).27-29
- Variable serum protein binding: Theoretically, only the unbound concentration of drug in blood is capable of establishing equilibrium with pharmacologic receptors, thus making it a better predictor of response than total drug concentration. Most drug concentrations in serum, plasma, or blood, however, are measured as the summed concentration of bound and unbound drug. It is very likely that some of the patients who show toxicity within the conventional therapeutic range have abnormally low protein binding and high concentrations of unbound drug in blood.30 Low protein binding of a drug in blood can be the result of either reduced protein concentrations or the presence of other substances in blood that displace the drug from protein binding sites.
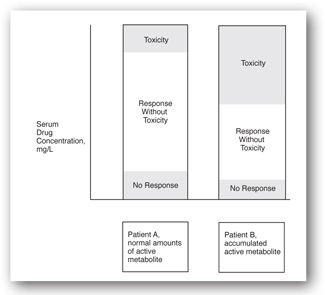
FIGURE 5-2. Representation showing how the individual therapeutic range of a hypothetical drug can differ in a patient with renal impairment because of accumulated active metabolite.
In summary, the therapeutic range reported by the laboratory is only an initial guide and is not a guarantee of desired clinical response in any individual patient. Every effort must be made to consider other signs of clinical response and toxicity in addition to the drug concentration measurement. Therapeutic ranges for the most commonly monitored drugs discussed in the Applications section of this chapter are reported in Table 5-3.
AIDS = acquired immune deficiency syndrome.
SAMPLE TIMING
Incorrect timing of sample collection is the most frequent source of error when therapeutic drug monitoring results do not agree with the clinical picture.19,31 Warner reviewed five studies in which 70% to 86% of the samples obtained for therapeutic drug monitoring purposes were not usable. In most cases, this was the result of inappropriate sample timing, including lack of attention to the time required to reach a steady-state. There are two primary considerations for sample timing: (1) how long to wait after initiation or adjustment of a dosage regimen, and (2) when to obtain the sample during a dosing interval.
At steady-state. When a drug dosage regimen (a fixed dose given at a regularly repeated interval) is initiated, concentrations are initially low and gradually increase until a steady-state is reached. Pharmacokinetically, steady-state is defined as the condition in which the rate of drug entering the body is equal to the rate of its elimination. For the purpose of therapeutic drug monitoring, a steady-state means that drug concentrations have leveled off at their highest and, when given as the same dose at a fixed interval, the concentration versus time profiles are constant from interval to interval. This is illustrated in Figure 5-3 for a constant infusion and a chronic intermittent dosage regimen.
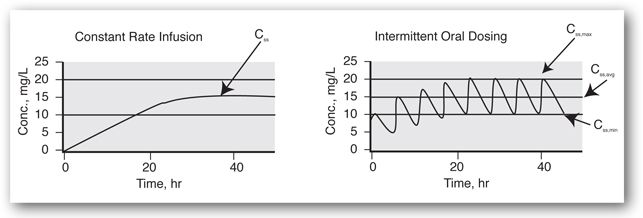
FIGURE 5-3. Concentration versus time plots for a constant infusion and intermittent therapy after initiation of therapy, without a loading dose. The half-life for this hypothetical drug is 8 hours. Thus, 88% of the eventual average steady-state concentration (Css,avg) is attained in 24 hours.
Drug concentration measurements should not be made until the drug is sufficiently close to a steady-state, so that the maximum benefit of the drug is ensured. (See Minicase 2.) The time required to reach a steady-state can be predicted if the drug’s half-life is known, as follows:
Importance of Documenting Drug Administration Times
ANGELA M. IS A 35-YEAR-OLD FEMALE who is receiving aminoglycoside monotherapy for treatment of a gram-negative infection. According to the medical chart, she has received 5 doses of tobramycin as 100 mg infused over 30 minutes, q 8 hr on a 6 a.m./2 p.m./10 p.m. schedule. The estimated half-life of tobramycin, based on Angela M.’s creatinine clearance, is 4.5 hours. Two serum tobramycin levels are ordered in order to determine if peak and trough serum levels are within the desired ranges of 6–10 mg/L and 0.5–2 mg/L, respectively. A trough serum level drawn at 1:50 p.m., just before the start of infusion of the 6th dose is 0.8 mg/L. A second level, drawn 30 minutes after the end of infusion of the 6th dose, is used to calculate a peak serum tobramycin level of 7.8 mg/L. Based on this information, the current regimen is continued. Repeat levels drawn 2 days later, however, reveal a peak tobramycin level of 10.1 mg/L and trough of 2.9 mg/L. Renal function, as indicated by creatinine clearance, has not changed in this patient. If this second set of serum concentrations is accurate, a dosage adjustment is critical to avoid aminoglycoside toxicity.
Question: What are the possible explanations for the apparent change in serum tobramycin level results? Which set of tobramycin levels accurately reflects the current dosage regimen?
Discussion: In a situation such as this, one should always consider whether the first levels were drawn at a steady-state. With an estimated tobramycin half-life of 4.5 hours, a steady-state was most certainly attained after 5 doses or 40 hours. A second consideration would be changing renal function. Since the aminoglycoside antibiotics are handled almost exclusively by the kidney, a decrease in renal function would explain the increase in serum tobramycin levels. In this case, however, renal function has not changed. A third consideration would be laboratory error or assay interference/artifact. Artifactually low serum tobramycin concentrations might have resulted if beta-lactam antibiotics had been coadministered during the time that the first set of samples was drawn; no other antibiotics were being coadministered, however. Assuming that laboratory error is ruled out, one must finally investigate the possibility of inaccurate documentation of either blood sample or drug administration times. Further scrutiny revealed that the 5th dose of tobramycin was held. Consequently, an extra 8 hours of washout occurred prior to blood sampling and the levels drawn before and after the 6th dose where lower than would be expected. These levels did not reflect the 100 mg, q-8-hr tobramycin regimen. After ensuring that all subsequent doses were appropriately administered and that the second set of blood samples was appropriately timed and documented, an adjusted dosage regimen of 80 mg q 12 hr was ordered.
| Number of Half-Lives | Percentage of Steady-State Attained |
| 2 | 75% |
| 3 | 88% |
| 4 | 94% |
| 5 | 97% |
This means the clinician should wait three half-lives at a minimum before obtaining a sample for monitoring purposes. The clinician should also anticipate that the “usual” half-life in a given patient may be actually longer due to impaired elimination processes, and it may be prudent to wait longer if possible. The half-lives of drugs that are typically monitored are reported in the Applications section, and typical times to steady-state are reported in Table 5-3.
Sometimes drugs are not given as a fixed dose at a fixed interval, or they may undergo diurnal variations in pharmacokinetic handling.32,33 While the concentration versus time profiles may differ from each other within a given day, the patterns from day-to-day will be the same if a steady-state has been attained. In cases of irregular dosing or diurnal variations, it is important that drug concentration measurements on different visits be obtained at similar times of the day for comparative purposes.
An unusual situation is caused by autoinduction, as exemplified by carbamazepine. The half-life of carbamazepine is longer after the first dose but progressively shortens as the enzymes that metabolize carbamazepine are induced by exposure to itself.34 The half-life of carbamazepine during chronic therapy cannot be used to predict the time required to reach a steady-state. The actual time to reach a steady-state is somewhere between the time based on the first-dose half-life and that based on the chronic-dosing half-life.
It is a common misconception that a steady-state is reached faster when a loading dose is given. While a carefully chosen loading dose will provide desired target levels following that first dose, the resulting level is only an approximation of the true steady-state level, and it will still require at least three half-lives to attain a true steady-state. Whenever possible, it is best to allow more time for a steady-state to be attained than less. This is also important because the average half-life for the population may not apply to a specific patient.
There are some exceptions to the rule of waiting until a steady-state is reached before sampling. If there is suspected toxicity early during therapy, a drug concentration measurement is warranted and may necessitate immediate reduction of the dose rate. Dosing methods designed to predict maintenance dosage regimens using pre-steady-state drug levels are useful when rapid individualization of the dosage regimen is needed.35-40
Within the dosing interval. Figure 5-3 shows typical concentration versus time profiles for a drug given by constant infusion and a drug given by oral intermittent dosing. Once a steady-state is attained, drug concentrations during a constant infusion remain constant, and samples for drug concentration measurements can be obtained at any time. When a drug is given intermittently, however, there is fluctuation in the drug concentration profile. The lowest concentration during the interval is known as the steady-state minimum concentration, or the trough. The highest concentration is known as the steady-state maximum concentration, or the peak. Also shown in Figure 5-3 is the steady-state average concentration (Css,avg), which represents the time-averaged concentration during the dosage interval. An important principle of dosing for drugs that show first-order behavior is that the average concentration during the interval or day will change in direct proportion to the change in the daily dose. This is covered in more detail in the Use of Levels for Dosage Adjustment section.
The degree of fluctuation within a dosing interval will depend on three factors: the half-life of the drug in that patient; how quickly the drug is absorbed (as reflected by the time at which a peak concentration occurs for that particular formulation); and the dosing interval. The least fluctuation (lowest peak:trough ratio) will occur for drugs with relatively long half-lives that are slowly absorbed or given as sustained-release formulations (prolonged peak time) and are given in divided doses (short dosing interval). However, drugs with relatively short half-lives that are quickly absorbed (or given as prompt-release products) and given only once daily will show the greatest amount of fluctuation within the interval.
The estimated degree of fluctuation (peak:trough ratio) for a given dosage regimen can be estimated by comparing the drug’s half-life in a patient to the difference between the dosing interval and the estimated time required to reach a peak concentration.41 The following guidelines may be used:
| (Interval-Peak Time)/Half-Life | Peak:Trough Ratio |
| 2.00 | 4.0 |
| 1.50 | 2.8 |
| 1.00 | 2.0 |
| 0.50 | 1.4 |
| 0.25 | 1.2 |
Using the information above, concentrations during the interval fluctuate very little (peaks are only 1.2 times troughs) if the difference between the dosing interval and peak time is one-quarter of the drug’s half-life. In that case, it may be assumed that concentrations obtained anytime during the interval are almost equivalent; the peak, trough, and average concentrations are roughly equal.
The choice of timing for samples within the dosing interval should be based on the clinical question to be addressed. Troughs are usually recommended for therapeutic confirmation, especially if the therapeutic range was formulated based on trough levels as for most of the antiepileptic drugs.14 Trough concentrations are also recommended if the indication for concentration monitoring is avoidance of inefficacy or distinguishing nonadherence from therapeutic failure. Trough concentrations should also be monitored if the patient tends to experience symptoms of inefficacy before the next dose (in which case a shortening of the dosing interval might be all that is needed). While it is logical to assume that the lowest concentration during the interval will occur immediately before the next dose, this is not always the case. Some products are formulated as delayed-release products (e.g., enteric-coated valproic acid) that are designed to be absorbed from the intestine rather than the stomach. As such, they may not begin to be absorbed for several hours after administration, and the level of drug from the previous dose continues to decline for several hours into the next interval. It is important to recognize that the predose level for those formulations is not the lowest level during the interval (Figure 5-4).
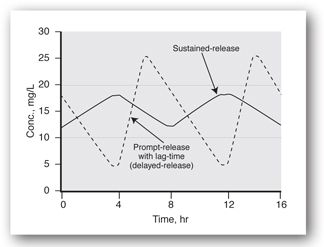
FIGURE 5-4. Concentration versus time profiles for a prompt-release formulation that exhibits a lag-time in its release or absorption (delayed-release) as compared to a sustained-release formulation without lag-time. Note that the lowest concentration during the dosing interval for the delayed-release product occurs at a time that is typically expected for the peak to occur.
Peak concentrations are monitored less often for drugs given orally because the time at which peak concentrations occur is difficult to predict. If a peak concentration is indicated, the package insert should be consulted for peak times of individual products. Peak concentration monitoring would be appropriate if the patient complains of symptoms of toxicity at a time believed to correspond with a peak concentration. Peaks may also be used for intravenous drugs (aminoglycosides and chloramphenicol) because the time of the peak is known to correspond to the end of the infusion. For the aminoglycosides, the peak level is believed to be a predictor of efficacy, while for chloramphenicol the peak concentration predicts both efficacy and adverse effects.42
Sometimes the clinician wishes to get an idea of the average level of drug during the day or dosing interval. This is particularly useful when the level is to be used for a dosage adjustment. Pharmacokinetically, the average level equals the area-under-the-curve (AUC) during the dosing interval (requiring multiple samples) divided by the interval. (Note: The AUC during an interval or portion of an interval is used as the monitoring parameter in place of single drug concentration measurements for certain drugs, such as some immunosuppressant and cytotoxic drugs, because it provides a better indication of overall drug exposure.) However, determination of the AUC, or Css,avg, by multiple sampling is not cost-effective for the most commonly monitored drugs. The following are alternatives to estimating the Css,avg without multiple samples:
- Look up the expected time to reach a peak concentration for the particular formulation and obtain a sample midway between that time and the end of the dosing interval.20
- Measure the trough level (as close to the time of administration of the next dose as possible) and use that along with the population value for the drug’s volume of distribution (Vd) to estimate the peak level as follows:
Peaksteady-state = (Dose/Vd) + measured trough
Then take the simple average of the trough and the peak to get an estimate of the average steady-state level.
- If you have reason to believe that there is very little fluctuation during the dosing interval, then a sample drawn anytime during the interval will provide a reasonable reflection of the average concentration.
There are special and extremely important timing considerations for some drugs, such as digoxin. It must reach specific receptors, presumably in the myocardium, to exhibit its therapeutic effect, but this takes a number of hours after the dose is administered. Early after a digoxin dose, levels in serum are relatively high, but response is not yet evident because digoxin has not yet equilibrated at its site of action. Thus, only digoxin levels that are in the postdistribution phase should be monitored and compared to the reported therapeutic range.
The timing of samples for other drugs may be based on the requirements for certain dosing methods. This is true for the aminoglycosides and certain lithium dosing methods. Sample timing for drugs like methotrexate will be specified in protocols because concentrations are used to determine the need for rescue therapy with leucovorin to minimize methotrexate toxicity.
While samples for drug concentration measurements may be preferred at certain times during a dosing interval, visits to physician offices often do not coincide with desired times for blood draws. One is then faced with the matter of how to interpret a level that is drawn at a time that happens to be more convenient to the patient’s appointment. The most critical pieces of information to obtain in this situation are (1) when the last drug dose was taken; (2) how compliant the patient has been; (3) timing of the sample relative to the last dose; and (4) the expected time of peak concentration. Some drugs are available as a wide variety of formulations (solutions, suspensions, prompt-release, and sustained- or extended-release solid dosage forms), and the package insert may be the best source of information for the expected peak time. Once again, drugs with relatively long half-lives given as sustained-release or slowly absorbed products in divided doses will have the flattest concentration versus time profiles, and levels drawn anytime during the interval are going to be similar. However, prompt-release drugs with short half-lives given less frequently will show more fluctuation. Knowing the expected peak time for the formulation in question is especially important for drugs that show more fluctuation during that interval. In that case, one can at least judge if the reported concentration is closer to a peak, an average (if midway between the peak and trough), or a trough.
SPECIMENS, COLLECTION METHODS, AND ASSAYS
Whole blood, plasma, and serum. Whole blood, plasma, serum, and ultrafiltrate of serum are commonly used specimens for drug concentration measurements. Unbound drug molecules in blood distribute themselves among red blood cells, binding proteins (albumin, alpha-1-acid glycoprotein [AAG], and lipoproteins) and plasma water based on how avidly they partition into red blood cells; the concentrations of binding proteins in blood; and the affinities of the binding proteins for the drug.30
Samples for plasma or whole blood analysis are collected in tubes that contain an anticoagulant. Plasma is created by centrifugation of the anticoagulated whole blood sample and collection of the upper layer containing plasma water, protein, unbound drug, and protein-bound drug. Samples for serum are collected in tubes without an anticoagulant and allowed to clot followed by centrifugation, while samples for unbound drug measurements, preferably serum, are ultrafiltered (as described below) to create serum water that contains only unbound drug. Concentrations of drug in ultrafiltrate of serum are always lower than the corresponding total (bound plus unbound) concentrations, especially for drugs that are highly bound to serum proteins. Because the only difference between plasma and serum is that the clotting factors have been consumed when serum is created, drug concentrations in plasma and serum are generally regarded to be equivalent. Concentrations of drug in whole blood are higher than the corresponding serum or plasma concentrations if the drug happens to concentrate within red blood cells.
The choice of serum, plasma, or whole blood for a drug concentration measurement depends in part on the requirements of the assay to be used. Serum or plasma would be preferred if hemoglobin interferes with the assay. An assay with marginal sensitivity might be more useful if whole blood is used for a drug such as cyclosporine that concentrates in the red blood cell. Some of the newer point-of-care methods have the advantage of using capillary whole blood from fingersticks, thus obviating the need for centrifugation or other sample preparation steps.
The choice of blood collection method is extremely important. For plasma analyses, it is important to know if the particular anticoagulant interferes with the assay, affects the stability or protein binding of the drug, or even dilutes the sample.43 There have been numerous reports that polymer-based gels, designed to form a barrier between serum and the clot during centrifugation, may also absorb drugs from the serum to varying extents. Technical improvements in these devices (known as serum separator tubes) have been made, but, nevertheless, several groups have categorically recommended that separation gels not be used for blood collection unless rigorous testing has first been done.44,45 Finally, there were a number of reports in the 1970s and 1980s of artifacts caused by contact of serum or plasma with rubber stoppers of evacuated blood collection tubes.46,47 Tris-(2-butoxyethyl)phosphate (TBEP) in the rubber stoppers leached into the serum or plasma and displaced basic drugs from AAG, thus causing redistribution of unbound drug into the red blood cells. When the samples were centrifuged, the total plasma or serum drug concentration was decreased to varying extents. Although recent studies of reformulated stoppers for these tubes show no artifacts, each laboratory must perform their own tests to ensure that similar artifacts are not observed.47
Specific recommendations for blood collection methods and collection tubes will be presented in the appropriate sections that follow for each drug or drug category.
Alternatives to blood sampling. Saliva has been proposed as a noninvasive alternative to blood sampling, particularly for children, for a number of drugs.48-50 Collection at home and mailing of samples may further offer convenience and cost savings.51 Saliva is a natural ultrafiltrate of plasma and, thus, may provide a closer reflection of the therapeutically active unbound drug concentration in serum as compared to the total drug concentration.52 Concentrations of drug in saliva are much lower than total concentrations in serum of highly bound drugs, and assay sensitivity should be considered if saliva samples are used for drug concentration monitoring.
Use of saliva drug concentration as a substitute for total or unbound drug concentration in serum or plasma requires that the saliva:plasma (S:P) drug concentration ratio be stable, at least within a patient. The S:P ratio, however, can be influenced by salivary flow rate, saliva pH, and contamination by residual drug in the mouth. With the exception of saliva pH, these factors can be controlled by careful selection of the collection method. The degree of a drug’s ionization affects the extent to which it passively diffuses from blood into saliva. Blood pH is relatively constant, but saliva pH varies widely during the day within a patient, and the S:P ratio may, therefore, change considerably during the day. Neutral drugs are not expected to show variable S:P ratios within patients, while acids or bases with pKa values similar to blood pH are most likely to be sensitive to variations in saliva pH. Correction of the S:P ratio may be possible, however, if the saliva pH is measured.52,53
Whole saliva is most often collected, and it can be either stimulated or unstimulated. Stimulated saliva is preferred: it produces a larger volume, minimizes the pH gradient between saliva and blood, and provides more stable S:P ratios. Methods for stimulating whole saliva production include the chewing or rolling of inert materials (paraffin, wax, and glass marble) in the mouth or applying a small amount of citric acid to the tongue.54 Novel methods have been proposed for collection of saliva for home monitoring purposes in children. One involves placing a gauze-wrapped cotton ball, with attached string, in the child’s mouth for a period of time and squeezing saliva from the retrieved cotton using a plastic syringe.55 Special devices that collect saliva from specific salivary glands are commercially available. While they offer the advantage of reduced viscosity, they must be checked to ensure stability and recovery of any absorbed drug.48,56 Regardless of the method used, contamination of saliva from residual drug in the mouth must be avoided by collecting saliva no earlier than 2–3 hours after a dose and rinsing the mouth with deionized water prior to collection.
Lacrimal fluid has been proposed as an ideal medium for monitoring unbound drug, since it does not have the problem with pH changes.57-59 Tears may be stimulated by exposing the eye to cigarette smoke or having the patient sniff formaldehyde fumes. In another method, tears are collected by hooking small strips of blotting paper over the lower lid of the eye for a few minutes.58 The major limitation for assay of drug in tears is the sensitivity of the assay, since large volumes cannot be collected.
Storage. The following factors can affect drug concentrations in serum, serum ultrafiltrate, plasma, whole blood, saliva, and lacrimal fluid: exposure to light, temperature, storage container, and presence of other drugs or endogenous substances.
Assays. Interpretation of any drug concentration measurement requires full knowledge of the strengths and limitations of the analytical method that was used. Laboratories should readily supply clinicians with all details of assay performance, including linearity, coefficients of variation at high and low concentrations, minimum detection limit, and potential interferences. While the laboratory must consistently conform to accepted standards for accuracy and reproducibility, requirements for assay sensitivity and specificity will depend on the intended use, the therapeutic range, the volume to be analyzed, and the nature of the specimen (serum/plasma, whole blood, serum ultrafiltrate, or saliva).
Knowledge of the sensitivity of a method (the ability to quantitate drug when drug is present) is helpful to interpret a laboratory report of nondetectable. If the method is highly sensitive, nondetectable may actually mean there is no drug in the sample. If the method is less sensitive, however, there could be drug in the sample but just not enough to register.60 The minimum detection limit will be of greatest concern for drugs with lower therapeutic ranges (i.e., in the mcg/L range) such as digoxin and the PIs. More sensitive assays are also required when there are small sample volumes, such as in neonates. Drug concentrations in saliva or in ultrafiltrates of serum may require more sensitive assays if the drugs are highly bound to serum proteins. In some cases, assays may be based on blood concentrations for drugs that concentrate in red blood cells in order to overcome limitations of sensitivity.61
Knowledge of the specificity (the ability to detect nothing when nothing is there) of a method is equally important. If the drug concentration report from the laboratory is higher than would be expected with no attendant toxicity, the clinician might suspect an interference with the assay. Substances that may interfere with assays include drug metabolites, other drugs, and endogenous substances (e.g., lipids, bilirubin, hemoglobin, or uremic byproducts that accumulate in renal impairment). As an example, endogenous digoxin-like immunoreactive substances accumulate in the serum of neonates, pregnant patients, and patients with liver or renal disease and are reported to cause falsely elevated digoxin concentration readings.62 Some assays are purposely developed to be nonspecific for purposes of drug abuse screens. Examples are the immunoassays used for detection of benzodiazepines, tricyclics, or barbiturates.
Knowledge of the condition or appearance of the sample at the time of assay may be important. A sample with a milky appearance may indicate lipemia; one with a dark yellow to gold appearance may mean high bilirubin levels; one with a pinkish tinge may mean hemolysis. All of these conditions can result in either overreading (positive bias) or underreading (negative bias) of a drug concentration, depending on the analytical method. Drugs that are more concentrated in red blood cells than in serum will have artifactually high-serum concentration readings in a hemolyzed sample, while drugs that are less concentrated in the red blood cell will dilute the serum or plasma resulting in artifactually low readings.
High-performance liquid chromatography (HPLC) and gas-liquid chromatography (GLC) are still used in clinical laboratories and are considered in many cases to be the reference methods. Homogenous immunoassays (fluorescence polarization immunoassay [FPIA], enzyme-multiplied immunoassay [EMIT], and cloned enzyme-donor immunoassay [CEDIA]) have become the methods of choice, however, because of ease of use, ability to automate, and rapid turnaround time. These immunologic methods are generally specific for the parent drug, but in some cases metabolites or other drug-like substances are recognized by the antibody.63 Certain drugs are not suitable for immunologic assays. Lithium, an electrolyte, is an example of this and must be analyzed using ion-selective electrode (ISE) technology, atomic absorption spectroscopy, or flame emission photometry.
The increased interest in drug assay methods for use in ambulatory settings, such as physician offices, has led to the development of immunoassay systems purported to be fast, reliable, and cost-effective.5,64-67 Most of these point-of-care testing methods have the capacity to produce results within 1–5 minutes; some use whole blood. Prior to 1988, less than 10% of all clinical laboratories were required to meet quality standards. Growing concern about lack of quality control in settings such as physician offices led to adoption of the 1988 Clinical Laboratory Improvement Amendments (CLIA) in which three levels of testing complexity were defined: waived, moderately complex, and highly complex. All drug concentration measurement testing is currently classified either as moderate or high complexity, and laboratories that perform these assays must maintain a quality control program, participate in proficiency testing programs, and be periodically inspected.5 These point-of-care testing methods vary in their reliability as compared to reference methods.68-71
USE OF LEVELS FOR DOSAGE ADJUSTMENT
A chronic intermittent dosage regimen has three components: the dose rate, the dosing interval, and the dose. For the dosage regimen of 240 mg q 8 hr, the dose is 240 mg, the interval is 8 hours, and the dose rate can be expressed as 720 mg/day or 30 mg/hr. The dose rate is important because it determines the average concentration (Css,avg) during the day. The degree of fluctuation within a dosing interval is highly influenced by the dosing interval.
Dosage adjustments for linear behavior. If a drug is known to have first-order bioavailability and elimination behavior after therapeutic doses, one can use simple proportionality to make an adjustment in the daily dose:
- If average levels are being monitored or estimated, one can predict that the average, steady-state drug concentration will increase in proportion to the increase in daily dose, regardless of any changes that were made in the dosing interval.
- If trough levels are monitored and the dosing interval will be held constant, the trough level will increase in proportion to the increase in daily dose.
- If trough levels are monitored for a drug that exhibits considerable fluctuation during the interval, and both the dose rate and dosing interval will be adjusted, the trough concentration will not be as easy to predict at a new steady-state and is beyond the scope of this chapter. If the trough concentration can be used to estimate the Css,avg, as described above, the Css,avg can be predicted with certainty to change in proportion to the change in daily dose.
Sampling after a dosage regimen adjustment, if appropriate, should not be done until a new steady-state has been reached. For a drug with first-order behavior, this should take the same period of time (three half-lives at a minimum) that it did after initiation of therapy with this drug.
Dosage adjustments for drugs with nonlinear behavior. All drugs will show nonlinear elimination behavior if sufficiently high doses are given. Some drugs, however, show pronounced nonlinear (Michaelis-Menten) behavior following doses that produce therapeutic drug concentrations. This means that an increase in the dose rate of the drug will result in a greater-than-proportional increase in the drug concentration. Phenytoin is an example of a drug with this behavior. Theophylline, procainamide, and salicylate also show some degree of nonlinear behavior but only at the higher end of their therapeutic ranges (and not enough to require special dosing methods).
Methods have been described to permit predictions of the effect of dose rate increases for phenytoin using population averages or actual measurements of the parameters that define nonlinearity, namely Vmax (maximum rate of metabolism) and Km (the “Michaelis constant”), but they are beyond the scope of this chapter.72 The most important rule to remember for dosage adjustments of drugs like phenytoin is to be conservative; small increases in the dose rate will produce unpredictably large increases in the serum drug concentration. It must also be remembered that the half-life of a drug like phenytoin will be progressively prolonged at higher dose rates. Increases in dose rate will require a longer period of time to reach a steady-state as compared to when the drug was first initiated.
Population pharmacokinetic or Bayesian dosage adjustment methods, which involve the use of statistical probabilities, are preferred by many for individualization of therapy.40,73 They are useful for drugs with both linear and nonlinear behavior.
PROTEIN BINDING, ACTIVE METABOLITES, AND OTHER CONSIDERATIONS
Altered serum binding. Total (unbound plus protein-bound) drug concentrations measured in blood, serum, or plasma are almost always used for therapeutic drug monitoring, despite the fact that unbound drug concentrations are more closely correlated to drug effect.30 This is because it is easier to measure the total concentration and because the ratio of unbound to total drug concentration in serum is usually constant within and between individuals. For some drugs, however, the relationship between unbound and total drug concentration is extremely variable among patients, or it may be altered by disease or drug interactions. For drugs that undergo concentration-dependent serum binding, the relationship between unbound and total concentration varies within patients. In all of these situations, total drug concentration does not reflect the same level of activity as with normal binding and must be cautiously interpreted because the usual therapeutic range will not apply. (See Minicase 3.)
Value of Unbound Antiepileptic Drug Serum Levels
FRANK S., A 66–YEAR-OLD CAUCASIAN MALE, was admitted to the emergency department (ED) with a chief complaint of vomiting, diarrhea, blurred vision and unsteady gait. He was diagnosed with epilepsy 3 years ago and was taking the following oral antiepileptic drugs at home: valproic acid 1000 mg BID; sodium phenytoin 200 mg TID; carbamazepine 300 mg BID; and levetiracetam 400 mg at bedtime. The patient also had a past medical history of hypertension and hyperlipidemia and was taking aspirin 81 mg every day, simvastatin 40 mg at bedtime, and metoprolol 100 mg BID. Physical examination in the ED revealed bilateral nystagmus and significant ataxia. The clinical picture was deemed consistent with antiepileptic drug toxicity and total serum concentrations of three of the antiepileptic drugs were ordered:
| Carbamazepine: | 6.4 mg/L (reference range: 4–12 mg/L) |
| Phenytoin: | 9.3 mg/L (reference range 10–20 mg/L) |
| Valproic acid: | 72 mg/L (reference range 50–100 mg/L) |
Free levels of the same three drugs were subsequently determined:
| Carbamazepine: | 1.9 mg/L (reference range: 1–3 mg/L) |
| Phenytoin: | 1.3 mg/L (reference range 1–2 mg/L) |
| Valproic acid: | 13.4 mg/L (reference range 2.5–10 mg/L) |
The valproic acid was held for 24 hours and then reintroduced at a dose rate of 250 mg TID. His symptoms resolved within 24 hours. A repeat unbound serum valproic acid level 1 week later was 5.2 mg/L.
Questions: How were total levels of antiepileptic drug misleading in this patient? How might sole reliance on total antiepileptic serum concentrations have led to a different clinical decision and outcome? How did free serum concentration monitoring aid in understanding the cause of the patient’s signs and symptoms?
Discussion: Frank S. had total serum concentrations of carbamazepine and valproic acid within the reference ranges for total concentrations of these drugs, while the total level of phenytoin was slightly below the lower limit of the laboratory’s reference range. Based solely on these total drug concentrations, the unaware clinician would be tempted to seek alternative explanations and likely delay the resolution of the patient’s signs and symptoms. Even worse, the clinician might be tempted to increase the daily dose of sodium phenytoin in an attempt to get the total phenytoin level to within the usual reference range for phenytoin. Measurement of free levels of these anticonvulsants revealed that the patient was probably getting appropriate daily doses of carbamazepine and phenytoin but was clearly receiving too much valproic acid. This was confirmed when the signs and symptoms resolved after reduction of the valproic acid dose rate and a repeat unbound serum valproic acid level at a new steady-state was within the laboratory’s reference range for free valproic acid.
Explanations for the “supratherapeutic” free valproic acid level in face of a “therapeutic” total level in this patient may include one or more of the following: (1) saturable (nonlinear) protein binding of valproic acid to albumin at higher dose rates of valproic acid; (2) inhibition of valproic acid metabolism by salicylic acid; and (3) displacement of valproic acid from albumin by phenytoin and/or salicylic acid. As a result, total valproic acid concentrations no longer reflect what is happening to the free, active valproic acid moiety, and the usual therapeutic reference range of total concentrations cannot be used.
Minicase 3 is adapted from reference 142.
The direct measurement of unbound drug concentration would seem to be appropriate in these situations. Drugs for which total concentration monitoring is routinely performed (but for which unbound concentration monitoring has been proposed) include carbamazepine, disopyramide, lidocaine, phenytoin, quinidine, and valproic acid. Of these, correlations between unbound drug concentration and response have been only weakly established for carbamazepine and disopyramide but more firmly established for phenytoin.26,30,74
Unbound drug concentration measurements involve an extra step prior to analysis—separation of the unbound from the bound drug. Equilibrium dialysis and ultracentrifugation may be used in a research setting, but ultrafiltration is the method of choice for a clinical laboratory.30,75 Commercial systems for unbound drug concentration measurements involve centrifugation of serum in tubes containing a semipermeable membrane (e.g., Millipore Centrifree® UF device). The ultrafiltrate, containing unbound drug, is collected in a small cup and assayed. The method used for analysis of the ultrafiltrate must be sufficiently sensitive since lower drug concentrations will be observed for highly bound drugs. Specificity of the assay may also be especially important. The ratio of metabolite to parent drug is likely to be greater in the ultrafiltrate since most metabolites are not as highly bound to protein as the parent. Thus, an immunoassay that shows acceptable specificity using total serum might show unacceptable specificity using ultrafiltrate.76
If unbound drug concentration measurements are unavailable, too costly, or considered impractical, the following alternative approaches to interpreting total drug concentrations in situations of altered serum protein binding may be used:
- Use of equations to normalize the measured total concentration: Sheiner and Tozer were the first to propose equations that can be used to convert a measured total concentration of drug (phenytoin in this case) to an approximation of what the total level would be if the patient had normal binding.77 Equations to normalize total phenytoin concentrations have been used for patients with hypoalbuminemia, impaired renal function, and concurrent valproic acid therapy.78 Once the total level has been normalized, it may be compared to the conventional therapeutic range. It must be noted that this normalization method may not be a reliable substitute for measurement of the unbound phenytoin level.
- Normalize the measured total concentration using literature estimates of the abnormal unbound drug fraction: An alternative method for normalizing the total concentration can be used if reasonable estimates of the abnormal and normal unbound reactions of the drug can be ascertained (i.e., from the literature). The normalized total concentration (Cnormalized) can be estimated as

where Cmeasured is the measured total concentration reported by the laboratory.
- Predictive linear regression equations: Some studies have reported the ability to predict unbound drug concentrations in the presence of displacing drugs if the total concentrations of both drugs are known. This has been done to predict unbound concentrations of phenytoin and carbamazepine, both in the presence of valproic acid.79,80 These unbound drug concentrations should be compared to corresponding therapeutic ranges of unbound drug, which can be estimated for any drug if the normal unbound fraction and the usual therapeutic range of total concentrations (TR) are known:
TRu = TR × normal unbound fraction
- Use of saliva or tears as a substitute for unbound drug concentration: This may be a reasonable alternative so long as studies have shown a strong correlation between unbound concentrations in serum and concentrations in saliva or lacrimal fluid. The concentration of drug in saliva or tears may not be equal to the concentration in serum ultrafiltrate. Therefore, the laboratory should have determined a reliable conversion factor for this. The calculated unbound concentration may then be compared to the estimated therapeutic range for unbound concentrations as described above.
Active metabolites. Interpretation of parent drug concentration alone, for drugs with active metabolites that are present to varying extents, is difficult at best. Active metabolites may contribute to therapeutic response, to toxicity, or to both. Since metabolites will likely have different pharmacokinetic characteristics, they will be affected differently than the parent drug under different physiologic and pathologic conditions.
For drugs like primidone (metabolized to phenobarbital) and procainamide (metabolized to NAPA), the laboratory will typically report both the parent drug and the metabolite as well as a therapeutic range for both. While a therapeutic range for the sum of procainamide and NAPA may be reported by some laboratories, this practice is discouraged since the parent and metabolites have different types of pharmacologic activities.
Enantiomeric pairs. Some drugs exist as an equal mixture (racemic mixture) of enantiomers, which are chemically identical but are mirror images of each other. Because they can interact differently with receptors, they may have very different pharmacodynamic and pharmacokinetic properties. The relative proportions of the enantiomers can differ widely among and within patients. Thus, a given concentration of the summed enantiomers (what is routinely measured using achiral methods) can represent very different activities.
Table 5-3 provides relevant information about protein binding, active metabolites, enantiomers, and other influences on serum concentration interpretation for drugs discussed in the Applications sections that follow.
APPLICATIONS
Analgesic/Anti-inflammatory Drugs
Salicylic Acid
Therapeutic range. Salicylic acid is used to reduce fever and relieve pain and inflammation associated with a variety of conditions. The therapeutic range for the analgesic and antipyretic effects of salicylic acid is commonly reported as 20–100 mg/L.81,82 Salicylate is more commonly monitored, however, for its anti-inflammatory effect: while the commonly reported therapeutic range is 100–250 mg/L, effective concentrations may be as low as 70 mg/L and as high as 300 mg/L.81,82 The concentrations associated with toxicity can overlap considerably with those associated with efficacy. Tinnitus, for example, may be experienced at concentrations as low as 200 mg/L. Indications for monitoring salicylate concentrations, other than suspected overdose or chronic salicylate abuse, include suspected toxicity; suspected nonadherence; change in renal function, mental status, acid–base balance, or pulmonary status; and anticipated drug–drug interactions.
Sample timing. Salicylic acid undergoes nonlinear elimination, and, thus, the half-life progressively increases from 3–20 hours as drug accumulates to within the range of 100–300 mg/L.83 Because of the progressive prolongation of half-life during initiation of therapy, samples for salicylate monitoring should not be obtained earlier than after 1 week of therapy.84 The rate of salicylate absorption, while usually fast, is slowed during food intake or when enteric-coated formulations are administered.84 Trough samples are generally advised for purposes of therapeutic drug monitoring, as they are the most reproducible.84 Timing of the sample within the interval was not deemed critical in patients with juvenile rheumatoid arthritis who are dosed using an interval of 8 hours or less.85
Specimens, collection methods, and assays. Blood samples for determination of salicylate concentration should be collected in tubes without additives or in tubes containing heparin or ethylenediaminetetraacetic acid (EDTA).81 Recent studies of certain evacuated serum separator tubes show they are also acceptable for blood collection for salicylate monitoring.44,86 Saliva concentrations are extremely variable when compared to unbound salicylate concentrations, and the variability is not explained by pH alone.50 Salicylate in serum may be stored refrigerated for up to 2 weeks.81
Colorimetric methods are used for salicylate determination as well as GLC, HPLC, and FPIA. The FPIA method performs exceptionally well and is recommended over colorimetric methods especially for icteric serum or plasma.87,88 Saliva is proposed to be a reasonable alternative to icteric serum, however, if a colorimetric method must be used.81 An immunoassay-based point-of-care method has been developed to simultaneously screen for salicylate and acetaminophen overdose.89
Use of levels for dosage adjustment. Two of the metabolic pathways for salicylate are capacity-limited, such that increases in dose rate produce greater-than-proportional increases in unbound serum drug concentrations and response. Because there is also concentration-dependent serum protein binding, total concentrations may mask this nonlinear relationship between dose rate and unbound drug concentration. Droomgoole and Furst provide an algorithm for adjustment of salicylate doses based on total serum salicylate levels.82
Protein binding, active metabolites, and other considerations. The binding of salicylate to albumin is concentration-dependent. Specifically, it is approximately 90% bound at total concentrations of 100 mg/L and decreases to 76% bound at levels as high as 400 mg/L.82 The unbound fraction of salicylate is known to increase during pregnancy and in patients with nephrotic syndrome, liver disease, and uremia.82 While salicylate would seem to be an ideal candidate for unbound concentration monitoring, a therapeutic range for unbound salicylate has not been established. Nevertheless, the clinician should be cautious that total concentrations within the usual therapeutic range may be associated with toxic responses in patients who are suspected to have abnormally low serum binding. No significant differences in the unbound percentage of salicylate in serum were observed among patients with juvenile rheumatoid arthritis, despite widely variable albumin concentrations, suggesting that total concentration monitoring is more appropriate in this group.90
Antiasthmatics
Theophylline
Therapeutic range. Some clinicians still use 10–20 mg/L as the accepted therapeutic range for theophylline for management of acute bronchospasm associated with asthma and chronic obstructive pulmonary disease.91 The 2007 NIH Expert Panel Report, Guidelines for the Diagnosis and Management of Asthma, stipulates, however, a more conservative range of 5–15 mg/L.92 Most patients will respond at serum concentrations within this range, but levels as low as 2 mg/L may provide anti-inflammatory effects in some patients, while levels up to 20 mg/L may be necessary in others.91,93 There is an 85% probability of adverse effects with levels above 25 mg/L, and levels above 30–40 mg/L can be associated with dangerous adverse events.94 Adverse effects typically experienced by adults include nausea, vomiting, diarrhea, irritability, and insomnia at levels above 15 mg/L; supraventricular tachycardia, hypotension, and ventricular arrhythmias at levels above 40 mg/L; and seizures, brain damage, and even death at higher levels. It must also be noted that side effects such as nausea and vomiting, while common, do not occur in all patients and should never be considered prodromal to the occurrence of the more serious side effects.95
Theophylline is also indicated for treatment of neonatal apnea, although caffeine is usually preferred.96 The therapeutic range in neonates is generally considered to be 5–10 mg/L but may be as low as 3 mg/L on the low end to 14 mg/L on the high end.91,97-99 Adverse effects in neonates include lack of weight-gain, sleeplessness, irritability, diuresis, dehydration, hyperflexia, jitteriness, and serious cardiovascular and neurologic events.94 Tachycardia has been reported in neonates with levels as low as 13 mg/L.100
In summary, there is considerable overlap of therapeutic and toxic effects within the usual therapeutic ranges reported for theophylline in neonates, children, and adults. Therefore, serum concentrations should never be interpreted in the absence of information about the patient’s clinical status. Indications for theophylline monitoring include therapeutic confirmation of effective levels after initiation of therapy or a dosage regimen adjustment, anticipated drug–drug interactions, change in smoking habits, and/or changes in health status that might affect the metabolism of theophylline.
Sample timing. The half-life of theophylline is greatly affected by age, disease, concurrent drugs, smoking, and any physiologic condition that affects its metabolism. The half-life of theophylline can range anywhere from 3–5 hours in children or adult smokers to as long as 50 hours in nonsmoking adults with severe heart failure or liver disease.91 Steady-state will be reached in 24 hours for the average patient with an elimination half-life of 8 hours but will require much longer for patients with heart failure or liver disease (or for patients who are taking drugs known to inhibit theophylline metabolism). The time to steady-state in premature neonates may be as long as 9 days.97
The fluctuation of theophylline concentrations within a steady-state dosing interval can be quite variable—depending not only on the frequency of administration, type of formulation (sustained- or prompt-release), and half-life—but also on whether or not the dose was taken with a meal.94 There are many theophylline formulations available. Thus, it is important to consult the product information to determine the anticipated peak times. For prompt-release formulations, peak times are 1–2 hours; peak times for sustained-release formulations occur later and are difficult to predict.95
Trough concentrations of theophylline are most reproducible and should always be obtained if at all possible. Comparisons of trough levels from visit to visit will also be facilitated if samples are obtained at the same time of day on each visit. This is because of diurnal variations in the rate of theophylline absorption.94
Specimens, collection methods, and assays. Plasma or serum is used for most assays; whole blood may be used in some of the point-of-care systems.94 There are no particular concerns about blood collection tubes. Prolonged storage in red-top evacuated tubes or serum separator tubes had no effect on theophylline concentrations in serum.44
Many studies suggest saliva theophylline concentrations to be reliable predictors of total or unbound theophylline concentrations in serum or plasma.56,101,102 Both unstimulated and stimulated saliva were equally good predictors of theophylline concentrations in serum in one study.101 Either citric acid or the chewing of Parafilm® may be used for stimulation of whole saliva production.102 A study of an oral mucosal transudate collection device showed that once the S:P ratio was established for a given patient, saliva samples collected at home by the patient are reliable predictors of serum theophylline concentrations.56
While theophylline is often measured using HPLC, the most common assays for point-of-care methods and in clinical laboratories are based on FPIA or EMIT. The immunoassay methods offer acceptable sensitivity but may not be suitable for patients with renal failure who have accumulated theophylline metabolites.103,104 Caffeine and theobromine have been reported to interfere with theophylline measurements by some point-of-care methods. The Abbott Vision® system showed no interferences by bilirubin and triglycerides, but hemolyzed samples gave lower readings.68,105
Use of levels for dosage adjustment. Theophylline is usually assumed to undergo first-order elimination, but some of its metabolic pathways are nonlinear at concentrations at the higher end of the therapeutic range.94 The clearance of theophylline decreases by 20% as daily doses are increased from 210 mg to 1260 mg.94 While the magnitude of this nonlinear behavior does not require special methods for dosing, the clinician should expect somewhat greater-than-proportional increases in serum theophylline concentration with increases in dose rate, particularly as concentrations get into the upper end of the therapeutic range.
Protein binding, active metabolites, and other considerations. Theophylline is 35% bound to serum proteins in neonates and 40% to 50% bound to serum proteins in adults. Therefore, significant alterations in serum protein binding are unlikely.94 Theophylline is metabolized to the active metabolite caffeine, which is of minor consequence in adults. Caffeine concentrations in the serum of neonates who are receiving theophylline, however, are approximately 30% of theophylline concentrations and therefore contribute to the effect of theophylline during treatment of neonatal apnea. This may account for the slightly lower therapeutic range of theophylline in neonates as compared to adults. There are many other metabolites of theophylline, none of which possess significant activity.
Caffeine
Therapeutic range. Caffeine is indicated for neonatal apnea (apnea of prematurity) and is recommended over theophylline because it can be given once daily and is considered to have a wider therapeutic range.96 Concentrations as low as 5 mg/L may be effective, but most pediatric textbooks consider 10 mg/L to be the lower limit of the therapeutic range.99,106 Most clinicians consider 20 mg/L to be the upper limit of the range, and serious toxicity is associated with serum concentrations above 50 mg/L. Signs of toxicity include jitteriness, vomiting, irritability, tremor of the extremities, tachypnea, and tonic-clonic movements. Serum concentration measurements of caffeine may not be routinely necessary for apnea of prematurity in neonates.107 Neonates who do not respond as expected or in whom there is recurrence of apnea after a favorable response may benefit, however.
Sample timing. The half-life of caffeine in preterm infants at birth is 65–103 hours.106 Thus, a loading dose is always administered to attain effective levels as soon as possible. The long half-life means that caffeine concentrations will not fluctuate much during the interval, even when caffeine is administered once daily. Sampling in the postdistribution phase is recommended, but at least 2 hours postdose.
Baseline levels of caffeine must be obtained prior to the first caffeine dose in the following situations: (1) if the infant had been previously treated with theophylline, since caffeine is a metabolite of theophylline; and (2) if the infant was born to a mother who consumed caffeine prior to delivery. Reductions in the usual caffeine dose will be necessary if predose caffeine levels are present.
Specimens, collection methods, and assays. Because of the limited blood volume in neonates, it is generally recommended that blood samples of 75 µL or less be used.99 Caffeine from blood samples is measured as either serum or plasma. Recommendations for collection tubes include evacuated tubes without additives or tubes containing EDTA. Refrigeration at 4oC for up to 24 hours is acceptable.106 Common assays for caffeine include HPLC, GLC, and EMIT. The immunoassay method was demonstrated to be unaffected by hemolysis, hyperbilirubinemia, and lipemia.108
Saliva concentrations have been recommended as a noninvasive alternative to blood sampling, which would be particularly helpful in this population.106 The reported S:P concentration ratio can vary depending on the methods used. Therefore, it is important that collection and assay methods be consistently used within a given institution. De Wildt et al. developed a novel saliva collection method in which a cotton swab with attached gauze was placed in the mouth of the neonate 5–10 minutes after a drop of 1% citric acid solution had been placed in the cheek pouch.106 Saliva concentrations measured by HPLC predicted plasma concentrations reliably. Other collection methods (no stimulation or citric acid placed on the gauze) did not predict plasma concentrations as well.106
Use of levels for dosage adjustment. There is no data to suggest that caffeine undergoes nonlinear elimination. Thus, dosage adjustments by proportionality are acceptable. Dosage adjustments for caffeine are complicated by the fact that a true steady-state is not reached for at least 4 days, so any adjustments should be conservative.
Protein binding, active metabolites, and other considerations. Caffeine is only 31% bound to serum proteins and has no active metabolites.106
Antiepileptics
The antiepileptics that have clearly defined therapeutic ranges should be routinely monitored. Because they are used as prophylaxis for seizures that may not occur frequently, it is particularly important that effective serum concentrations of these drugs be ensured early in therapy. Indications for monitoring antiepileptic drugs include14,109 (1) documentation of an effective steady-state concentration after initiation of therapy; (2) after dosage regimen adjustments; (3) after adding a drug that has potential for interaction; (4) changes in disease state or physiologic status that may affect the pharmacokinetics of the drug; (5) within hours of a seizure recurrence; (6) after an unexplained change in seizure frequency; (7) suspected dose-related drug toxicity; and (8) suspected nonadherence.
Carbamazepine
Therapeutic range. Carbamazepine is indicated for the prevention of partial seizures and generalized tonic-clonic seizures, and the treatment of pain associated with trigeminal neuralgia.91,110 Most textbooks report a therapeutic range of 4–12 mg/L. Concentrations above 12 mg/L are most often associated with nausea and vomiting, unsteadiness, blurred vision, drowsiness, dizziness, and headaches in patients who are taking carbamazepine alone.34 Patients taking other antiepileptic drugs such as primidone, phenobarbital, valproic acid, or phenytoin, however, may show these adverse effects at levels as low as 9 mg/L. For this reason, many clinicians use a more conservative target therapeutic range of 4–8 mg/L.110 Serious adverse reactions are seen at levels greater than 50 mg/L.109
Carbamazepine 10,11-epoxide is an active metabolite that can be present in concentrations containing 12% to 25% carbamazepine, but it is not routinely monitored along with the parent drug. A suggested therapeutic range for this metabolite, used at some research centers, is 0.4–4 mg/L.91
In addition to the usual indications for monitoring, it is important to monitor carbamazepine concentrations if the patient is switched to another formulation (e.g., generic), since the bioavailability may be different.109
Sample timing. Because carbamazepine induces its own metabolism, it is recommended that initial dose rates of carbamazepine be relatively low and gradually increased over a 3- to 4-week period.34 For maximal induction or deinduction to occur, 2–3 weeks may be required after the maximum dose rate has been attained. Thus, a total of 6–7 weeks may be required for a true steady-state to be reached after initiation of therapy. After any dose rate changes or addition/discontinuation of enzyme-inducing or inhibiting drugs, 2–3 weeks will be required to reach a new steady-state.91
A trough level is generally preferred if there is a choice. The absorption of immediate-release carbamazepine tablets from the gastrointestinal tract is relatively slow and erratic, reaching a peak between 3 and 8 hours after a dose.111 Extended-release formulations are even more slowly absorbed. If carbamazepine is administered every 6 or 8 hours, serum levels during the dosing interval will remain fairly flat, and all levels will be fairly representative of a trough concentration. Less frequent dosing will result in more fluctuation in which case the time of the level relative to the last dose should be documented for appropriate interpretation. Use of the extended-release formulation of carbamazepine will minimize fluctuations caused by diurnal variations.112 Nevertheless, it is recommended that samples on repeated visits always be obtained at the same time of the day for purposes of comparison.109
Specimens, collection methods, and assays. Either serum or plasma collected in EDTA-treated tubes is acceptable for total carbamazepine measurements. Oxalate and citrate were shown to cause significant negative interferences in the measurement of carbamazepine by an EMIT method and a GLC method.113 Studies of a new serum separator tube (SST II®, Becton-Dickinson) showed that serum carbamazepine concentrations were stable for 24 hours at room temperature.44 Saliva has been proposed as a convenient noninvasive alternative, especially for children and for home monitoring.48,52,114 If saliva is used, a standardized protocol for obtaining the specimen must be approved by the laboratory. Both the chewing of Parafilm® and stimulation by citric acid have been used successfully.52.115 Saliva collected within 2 hours of oral administration may be contaminated by residual drug in the mouth.34
The most common assays for total carbamazepine include a wide variety of immunoassays.111 Some of the immunoassays cross-react with the 10,11-epoxide metabolite.116 This can be a particular problem if saliva is measured, as the ratio of epoxide to parent drug is higher in saliva.115 The active carbamazepine 10,11-epoxide is generally not routinely measured separately, even though it has been shown to exhibit anticonvulsant activity. Assays for unbound carbamazepine, monitored rarely, are done by ultrafiltration followed by one of the other assay methods.30 Severe hemolysis may result in inaccurate measurement by the immunologic methods in which case one of the chromatographic methods is suggested.117
Use of levels for dosage adjustment. Carbamazepine exhibits first-order behavior following therapeutic doses. Thus, increases in dose rate will result in a proportional increase in the average steady-state level of carbamazepine. If the dose is adjusted without a change in the interval, a level drawn at the same time within the interval will increase in proportion to the increase in dose.
Protein binding, active metabolites, and other considerations. In most patients, carbamazepine is 70% to 80% bound to serum proteins, including albumin and AAG.118 In some patients, however, unbound percentages as low as 10% have been reported.91 Measurements of unbound carbamazepine concentrations are not generally recommended or necessary. Rather, total carbamazepine concentrations should be carefully interpreted in situations of suspected altered protein binding. Decreased binding might be anticipated in liver disease, hypoalbuminemia, or hyperbilirubinemia.91 Increased binding might be expected in cases of physiologic trauma due to elevated AAG concentrations, but this would be a rare occurrence. Valproic acid has been shown to displace carbamazepine from albumin; an equation was proposed to predict unbound carbamazepine concentrations in this situation.80 Correlations between saliva and unbound carbamazepine concentrations are strong.52 Thus, saliva sampling might be considered in situations of suspected alterations in carbamazepine binding.
Drug–drug interactions that are expected to result in a higher proportion of active 10,11-epoxide metabolite relative to the parent drug (e.g., concurrent phenytoin, phenobarbital, or valproic acid) may alter the activity associated with a given carbamazepine concentration. It is suggested that a lower therapeutic range of 4–8 mg/L be used when those drugs are given concurrently.14
Ethosuximide
Therapeutic range. Ethosuximide is indicated for the management of absence seizures. The therapeutic range is generally considered to be 40–100 mg/L.119 Eighty percent of patients will achieve partial control within that range, and 60% will be seizure-free. Some patients will require levels up to 150 mg/L.91 Side effects are usually seen at concentrations above 70 mg/L and include drowsiness, fatigue, ataxia, and lethargy.91 Ethosuximide does not require as much monitoring as some of the other antiepileptics, but it is important to ensure effective levels after initiation of therapy or a change in dosage regimen.
Sample timing. The half-life of ethosuximide is quite long—60 hours in adults and 30 hours in children.111 Thus, it is advised to wait as long as 1 week to 12 days before obtaining ethosuximide levels for monitoring purposes.14,119 While it is generally advised that trough concentrations be obtained, levels drawn anytime during the dosing interval should be acceptable because there will be very little fluctuation if ethosuximide is given in divided doses. Peak concentrations of ethosuximide administered as a capsule are attained in 3–7 hours.111,119
Specimens, collection methods, and assays. Ethosuximide is usually measured by immunoassay.111 Serum or plasma may be used for determination of ethosuximide concentrations. A variety of blood collection tubes have been tested, and none have interfered with measurement of ethosuximide.47 Ethosuximide does not bind to serum proteins. Therefore, measurement of unbound levels is never necessary. Studies have shown saliva ethosuximide concentrations to be equal to serum or plasma concentrations, thus making saliva a convenient alternative, especially in children.48,115
Use of levels for dosage adjustment. Ethosuximide is reported to display nonlinear elimination, but primarily at concentrations near the upper end of its therapeutic range. Somewhat greater-than-proportional increases in drug concentration with increases in doses can therefore be expected when higher dose rates are use.
Protein binding, active metabolites, and other considerations. Ethosuximide is negligibly bound to serum proteins and its metabolites have insignificant activity. While ethosuximide is administered as a racemic mixture, the enantiomers have the same pharmacokinetic properties. Thus, measurement of the summed enantiomers is acceptable.120
Phenobarbital/Primidone
Primidone and phenobarbital are both used for management of generalized tonic-clonic and partial seizures.91 Phenobarbital is used for febrile convulsions and hypoxic ischemic seizures in neonates and infants.107 Primidone is used for treatment of essential tremor in the elderly.109 Although primidone has activity of its own, most clinicians believe that phenobarbital—a metabolite of primidone—is predominantly responsible for primidone’s therapeutic effects. These two drugs will, therefore, be considered together.
Therapeutic ranges. The therapeutic range of phenobarbital for treatment of tonic-clonic, febrile, and hypoxic ischemic seizures is generally regarded as 10–40 mg/L, while concentrations as high as 70 mg/L may be required for refractory status epilepticus.109,121 Eighty-four percent of patients are likely to respond with concentrations between 10 and 40 mg/L.121 Management of partial seizures seems to require higher phenobarbital concentrations than management of bilateral tonic-clonic seizures.14 Concentrations of phenobarbital are always reported when primidone levels are ordered. The therapeutic range of primidone reported by most laboratories is 5–12 mg/L.14,109 Fifteen to 20% of a primidone dose is metabolized to the active phenobarbital; the side effects of primidone are mostly related to phenobarbital.91 Central nervous system side effects such as sedation and ataxia generally occur in chronically treated patients at phenobarbital levels between 35 and 80 mg/L. Stupor and coma have been reported at phenobarbital concentrations above 65 mg/L. 111,121
Sample timing. The half-life of phenobarbital is the rate-limiting step for determining the time to reach steady-state after primidone administration. The half-life of phenobarbital averages 5 days for neonates and 4 days for adults.121 Since phenobarbital or primidone dosage may be initiated gradually, steady-state is not attained until 2–3 weeks after full dosage has been implemented. Because phenobarbital has such a long half-life, levels obtained anytime during the day would provide reasonable estimates of a trough concentration. Ideally, levels should be obtained from visit to visit at similar times of the day.121
Specimens, collection methods, and assays. Serum or plasma is acceptable for measurements of phenobarbital and primidone; whole blood is generally used for point-of-care methods. Use of a new serum separator tube (SST II®, Becton-Dickinson) did not cause a problem with phenobarbital determinations.44 The partitioning of phenobarbital into saliva is pH-sensitive. However, some studies have shown acceptable correlations with or without pH correction.48 Saliva concentrations of primidone are particularly sensitive to saliva flow rate changes but show strong correlations with serum concentrations of primidone when standardized collection methods are used.48 The clinical utility of just measuring primidone concentration in saliva is questionable.
Chromatographic methods (GLC, HPLC) may permit simultaneous determination of both primidone and phenobarbital, but immunoassays are most commonly used.111 There is potential for cross-reactivity of the immunoassay methods with coadministered barbiturates.111 No interferences from endogenous substances or blood collection tube components were observed with one immunoassay method for phenobarbital.122,123
Use of levels for dosage adjustment. Phenobarbital and primidone exhibit first-order elimination behavior, thus, a change in the dose rate of either drug will result in a proportional change in the average, steady-state serum concentrations.111,121
Protein binding, active metabolites, and other considerations. Phenobarbital is approximately 50% bound to serum proteins (albumin) in adults; primidone is not bound to serum proteins.109,124 Thus, total concentrations of both drugs are reliable indicators of the active, unbound concentrations of these drugs. While primidone has an active metabolite, phenylethylmalonamide (PEMA), the contribution to activity is unlikely to be significant.
Phenytoin
Therapeutic range. Phenytoin is primarily used for treatment of generalized tonic-clonic and complex partial seizures.125 It may also be used in the treatment of trigeminal neuralgia and for seizure prophylaxis following neurosurgery.91,125 Studies have shown that serum or plasma concentrations of phenytoin between 10 and 20 mg/L will result in maximum protection from primary or secondary generalized tonic-clonic seizures in most adult patients with normal serum binding. Ten percent of patients with controlled seizures have phenytoin levels less than 3 mg/L, 50% have levels less than 7 mg/L, and 90% have levels less than 15 mg/L.14 Levels at the lower end of the range are effective for bilateral seizures, while higher concentrations appear to be necessary for partial seizures.14 The therapeutic range of total concentrations in infants is lower due to lower serum protein binding: 6–11 mg/L.109 Concentration-related side effects include nystagmus, central nervous system depression (ataxia, inability to concentrate, confusion, and drowsiness), and changes in mental status, coma, or seizures at levels above 40 mg/L.109 While mild side effects may be observed at concentrations as low as 5 mg/L, there have been cases in which concentrations as high as 50 mg/L have been required for effective treatment without negative consequences.126
Some clinicians have proposed that monitoring of phenytoin be limited to unbound concentrations, particularly in patients who are critically ill or likely to have unusual protein binding.74,127,128 Unbound phenytoin concentrations are more predictive of clinical toxicity than are total phenytoin concentrations in these individuals.129 The therapeutic range of unbound phenytoin levels is presumed to be 1–2 mg/L for laboratories that determine the unbound phenytoin fraction at 25oC and 1.5–3 mg/L if done at 37oC.109
Sample timing. The time required to attain a steady-state after initiation of phenytoin therapy is difficult to predict due to phenytoin’s nonlinear elimination behavior. While the T50% is approximately 24 hours (considering the average population Vmax and Km values when levels are between 10 and 20 mg/L), there can be extreme variations in these population values. Half-lives between 6 and 60 hours have been reported in adults.109 Thus, a steady-state might not be attained for as long as 3 weeks. Some clinicians advise that samples be obtained prior to steady-state (after 3–4 days) in order to make sure that levels are not climbing too rapidly.125 Equations have been developed to predict the time required to reach a steady-state once Vmax and Km values are known.72 It is important to recognize that the time required to reach a steady-state in a given patient will be longer each time the dose rate is further increased.
Most clinicians advise that trough phenytoin concentrations be monitored.109 Phenytoin is quite slowly absorbed so that the concentration versus time profile is fairly flat. This is especially true when oral phenytoin is administered 2 or 3 times per day. In this case, a serum phenytoin sample drawn any time during the dosage interval is likely to be close to a trough concentration. The greatest fluctuation would be seen for the more quickly absorbed products (chewable tablets and suspension) in children (who have a higher clearance of phenytoin) given once daily. In this case, it is particularly important to document the time of sample relative to the dose—to identify if the level is closer to a peak, a trough, or a Css,avg.
Specimens, collection methods, and assays. Serum or plasma is generally recommended for total phenytoin measurements. Blood collected for plasma should not be anticoagulated with citrate or oxalate because these anticoagulants have been reported to cause negative interferences with measurements of phenytoin using the EMIT method.109,113 Anticoagulation with heparin is also of concern since activation of lipoprotein lipases may increase free fatty acid concentrations and displace phenytoin from albumin.109 While serum separator tubes are generally not recommended, more recent studies with a serum separator tube, the SST II (Becton Dickinson) tubes, showed that serum phenytoin concentrations were stable for 24 hours at room temperature.44,109
Saliva has been proposed as a viable alternative to monitoring plasma phenytoin concentrations, especially for children.48,51,54,115 It has also been proposed as a useful specimen for monitoring unbound phenytoin concentrations, particularly in patients taking valproic acid concurrently.48 Successful results in infants and children have been shown when saliva is stimulated using a small amount of citric acid on the tongue.50 Since the S:P ratio is affected by salivary flow rate, it is particularly important that the saliva collection procedure be carefully standardized.48
Immunoassays are the most common methods for measurement of total phenytoin concentrations in serum or plasma.72 The metabolites of phenytoin do not contribute to antiepileptic activity, but certain immunologic methods may measure accumulated phenytoin metabolites in patients with renal impairment. Immunoassays that use monoclonal antibodies or HPLC would be appropriate alternative methods for samples from patients with renal impairment.76 Fosphenytoin, the phenytoin prodrug, interferes with commonly used immunoassay methods.72 For this reason, serum for phenytoin monitoring should not be obtained earlier than 4 hours after administration of fosphenytoin, at which time it has been maximally converted to phenytoin.130 Assays for unbound phenytoin are usually done using ultrafiltered serum followed by one of the other assay methods.76 The unbound phenytoin fraction is affected by temperature. Therefore, this variable must be controlled.109 Hemolysis and lipemia do not interfere with phenytoin measurements using an FPIA method.131
Use of levels for dosage adjustment. Phenytoin exhibits pronounced nonlinear behavior following therapeutic doses. Thus, increases in dose rate will produce greater-than-proportional increases in the average serum concentration during the dosing interval. Several methods, described elsewhere, use population and/or patient-specific Vmax and Km values to predict the most appropriate dose rate adjustment.72 The clinician must be aware that the size of phenytoin daily dose increases should typically not be greater than 30 or 60 mg using sodium phenytoin or 25 or 50 mg using the chewable tablets.
Protein binding, active metabolites, and other considerations. The metabolites of phenytoin have insignificant activity. Phenytoin binds primarily to albumin in plasma and the normal unbound fraction of drug in plasma of adults is 0.1.109,125 Lower serum binding of phenytoin is observed in neonates and infants and in patients with hypoalbuminemia, liver disease, nephrotic syndrome, pregnancy, cystic fibrosis, burns, trauma, malnourishment, AIDS, and advanced age.91,132 Concurrent drugs (valproic acid, salicylate, and other nonsteroidal anti-inflammatory drugs [NSAIDs]) are known to displace phenytoin.91 Thus, a total level of phenytoin that is within the range of 10–20 mg/L in these patients might represent an unbound level that is higher than 1–2 mg/L (the therapeutic range of unbound levels). A total concentration of phenytoin in this situation can be misleading. Several approaches can be used in these situations: (1) an unbound phenytoin level can be ordered, if available; (2) the patient’s unbound phenytoin level can be calculated by estimating the unbound fraction in the patient (using the literature) and multiplying that by the patient’s measured phenytoin level (the resulting unbound level should then be compared to 1–2 mg/L); or (3) special equations may be used to convert the total phenytoin level to what it would be if the patient had normal serum protein binding.
The following equation was developed to normalize phenytoin (PHT) levels in patients with hypoalbuminemia and/or renal failure91,125,133:
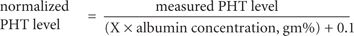
The value “X” is 0.2 for patients with low albumin and creatinine clearances equal to or above 25 mL/min and 0.1 mL/min for patients with normal or low albumin who are receiving dialysis. Total levels of phenytoin in patients with creatinine clearance values between 10 and 25 mL/min cannot be as accurately normalized; the clinical status of such patients should be carefully considered since total levels can be misleading. This equation for normalizing total phenytoin concentrations has been tested by groups of investigators in different groups of patients with mixed reviews; it is emphasized that it should be used only as a guide.
Valproic acid is known to increase the unbound fraction of phenytoin in serum.134 It has also been variably reported to inhibit the metabolism of phenytoin. These two occurrences together could mean that a level within the range of 10–20 mg/L is associated with adverse effects and an unbound phenytoin level greater than 2 mg/L. If unbound phenytoin concentrations are not available, the following equation—modified from its original form—may be useful to normalize the total phenytoin (PHT) level if the level of valproic acid (VPA) in that same sample has been measured78,125:
normalized PHT level = measured PHT level +
(0.01 × VPA level × measured PHT level)
Other equations have been used for estimating the unbound phenytoin concentration in the presence of valproic acid.135
Valproic Acid
Therapeutic range. Valproic acid is used for management of absence seizures, in addition to partial and generalized tonic-clonic and myoclonic seizures. It is also used for a variety of other conditions, including prophylaxis against migraine headaches and bipolar disorder.109 Most laboratories use 50–100 mg/L as the therapeutic range for trough total valproic acid concentrations. Some patients are effectively treated at lower levels, and others may require trough levels as high as 120 mg/L.136 Levels at the upper end of the therapeutic range appear to be necessary for treatment of complex partial seizures.136 The same therapeutic range has been used for patients with migraines or bipolar disorder, although the value of routine serum concentration monitoring for bipolar disorder has been questioned.137 The following concentration-related side effects may be seen: ataxia, sedation, lethargy, and fatigue at levels above 75 mg/L; tremor at levels above 100 mg/L; and stupor and coma at levels greater than 175 mg/L.81 The therapeutic range of total valproic acid concentrations is confounded by the nonlinear serum protein binding of this drug, which might explain some of the variable response among and within patients at a given total serum concentation.97
Sample timing. The half-life of valproic acid ranges between 7 and 18 hours in children and adults and 17 and 40 hours in infants.109 Thus, as long as 5 days may be required to attain a steady-state. The pattern of change in valproic acid concentrations varies from interval to interval during the day because of considerable diurnal variation.109,136 It is, therefore, recommended that samples always be obtained prior to the morning dose as this has been shown to be most consistent from day to day.136 Considerable fluctuation within the interval will be seen with the immediate-release capsule and syrup, which are rapidly absorbed. The enteric-coated, delayed-release Depakote® tablet displays a shift-to-the-right with respect to its concentration versus time profile, such that the lowest concentration during the interval may not be observed until 4–6 hours into the next dosing interval.111 It is important to know, however, that concentrations during the interval following administration of the enteric-coated tablet will show considerable fluctuation. The extended-release formulations (Depakote®ER and Sprinkle® capsules), if given in divided doses, provide less fluctuation in concentrations, and samples may be drawn at any time.
Specimens, collection methods, and assays. Serum or heparinized plasma are recommended; other anticoagulants may cross-react if immunoassay methods are used.109,113 Concentrations of valproic acid in saliva are very low and do not correlate well with plasma concentrations.48,54 Concentrations of valproic acid measured in tears collected using absorbent paper strips were shown to correlate well with unbound, valproic acid concentrations.58
Immunoassays are the most common methods for routine valproic acid serum concentration determinations.111 Unbound concentrations of valproic acid in ultrafiltrates of serum have been measured by immunoassay or HPLC.138
Use of levels for dosage adjustment. The metabolism of unbound valproic acid is linear following therapeutic doses. Thus, unbound valproic acid levels will increase in proportion to increases in dose rate.109,111 Because valproic acid shows nonlinear, saturable protein binding in serum over the therapeutic range, however, total concentrations will increase less than proportionally. This is important to keep in mind when interpreting total valproic acid levels.
Protein binding, active metabolites, and other considerations. Valproic acid is 90% to 95% bound to albumin and lipoproteins in serum. The unbound fraction of valproic acid shows considerable interpatient variability. It is increased in neonates, in conditions in adults associated with hypoalbuminemia (e.g., liver disease, nephrotic syndrome, cystic fibrosis, burns, trauma, malnutrition, and advanced age) and as a result of displacement by endogenous substances (e.g., bilirubin, free fatty acids, and uremic substances in end-stage renal disease) and other drugs (e.g., salicylate and other NSAIDs).91, 139,140 The increase in the unbound fraction of valproic acid during labor is believed to be the result of displacement by higher concentrations of free fatty acids.141 Valproic acid also shows intrapatient variability in the unbound fraction due to nonlinear binding. The unbound fraction of valproic acid is fairly constant at lower concentrations but progressively increases as total concentrations rise above 75 mg/L.109 Thus, total concentrations do not reflect unbound concentrations at the upper end of the therapeutic range. A therapeutic range for unbound valproic acid concentrations can be only approximated; assuming unbound fractions of 0.05–0.1 and a therapeutic range of total valproic acid concentrations of 50–100 mg/L, an unbound therapeutic range of 2.5–10 mg/L can be deduced.
Other Antiepileptic Drugs
Routine serum concentration monitoring is not recommended for most of the newer antiepileptic drugs.143,144 Lamotrigine, levetiracetam, felbamate, oxcarbazepine, tiagabine, topiramate, and zonisamide, however, have characteristics that might make concentration monitoring helpful for guiding therapy in certain situations or in special populations.22,145 While seizure control is associated with a wide range of gabapentin serum concentrations, some feel that its dose-dependent bioavailability may make serum concentration monitoring justified in some cases. Vigabatrin serum levels do not correlate with clinical effect because of its unusual mechanism of action—irreversible binding to an enzyme. Because severe adverse reactions observed in the postmarketing period have resulted in severe restrictions of felbamate use, it will not be discussed here.
Lamotrigine. The considerable pharmacokinetic variability among patients taking lamotrigine, due in part to significant drug–drug interactions, makes it a good candidate for therapeutic drug monitoring.143,146 The therapeutic range of lamotrigine was originally defined as 1–4 mg/L, but more recently 2.5–15 mg/L has been proposed.22,144,147 Some patients may tolerate concentrations above 20 mg/L.148 It has been suggested that concomitant therapy with other antiepileptics may alter the response to lamotrigine or its side effect profile at a given lamotrigine serum concentration.146 The half-life of lamotrigine can range from 15–30 hours on monotherapy.143 Thus, one should wait at least 1 week before obtaining samples after initiating or adjusting lamotrigine therapy.146 This drug exhibits linear pharmacokinetics; therefore, dose rate adjustments will result in proportionate changes in average serum concentrations. Because it is only 55% bound to serum proteins, measurements of unbound lamotrigine levels in serum are not necessary. Saliva concentrations of lamotrigine may be a useful alternative to blood sampling.143,149 There are HPLC assays currently available for lamotrigine.147 Routine lamotrigine monitoring is generally not recommended, but serum concentrations might be helpful in special populations or to determine if lack of response is related to unusually low levels or nonadherence.145,147
Levetiracetam. Serum concentration monitoring of levetiracetam is more important in pregnancy and in infants and children because of the higher clearances in these patients.22,145 The half-life ranges from 6–8 hours in adults and a steady-state should be attained within a week.144 Serum concentrations between 6 and 20 mg/L appear to be associated with response in most patients. Serum protein binding of levetiracetam is less than 10%, obviating the need for measurement of unbound levetiracetam concentrations.144
Oxcarbazepine. The pharmacologic effect of oxcarbazepine is primarily related to serum concentrations of its active monohydroxy metabolite (MHD). A therapeutic range of 13 to 35 mg/L for MHD may be used as a rough guide.22,144 The elimination half-life of MHD is quite variable, ranging from 7–20 hours, and is prolonged in renal impairment. The serum protein binding of MHD is low at 40%; therefore, unbound concentration monitoring of MHD is unnecessary. Routine monitoring of MHD is not warranted, but may be useful in patients with extremes of age or renal impairment, during pregnancy, or to rule out medication nonadherence.150
Tiagabine. Tiagabine shows pronounced interpatient pharmacokinetic variability. Trough levels between 20 and 100 mcg/L are associated with improved seizure control, but there is wide variation in response at any given total concentration.22,143,144 This could, in part, be due to variable serum binding (96% bound in serum on average). Valproic acid, salicylate, and naproxen have been shown to displace tiagabine from serum proteins.143,144 Tiagabine half-life ranges from 5–13 hours and may be even shorter in the presence of enzyme-inducing drugs.144,146 Tiagabine shows linear elimination behavior following therapeutic doses.
Topiramate. Topiramate levels are particularly influenced by interactions with other drugs, with levels as much as twofold lower when enzyme-inducing drugs are administered concurrently.151 The half-life ranges from 18–23 hours, and it has linear elimination behavior.143,144,146 Topiramate is less than 40% bound to serum proteins but shows saturable binding to red blood cells, thus suggesting that whole blood might be a preferable specimen for monitoring.144,146 Effective serum levels are generally reported to be between 5 and 20 mg/L with most patients responding at levels below 20 mg/L.22,143,144 No active metabolites have been identified. An FPIA assay method has been developed.146
Zonisamide. The pharmacokinetics of zonisamide are variable among patients and also highly influenced by interactions with other drugs.143 Zonisamide is approximately 40% bound to serum albumin, and, like topiramate, shows saturable binding to red blood cells, suggesting that whole blood monitoring might be preferable.144,146 The half-life is 50–70 hours but may be as short as 25 hours when enzyme inducers are coadministered.143 There are some reports suggesting nonlinear behavior at higher doses. The serum concentration range associated with response is 10–38 mg/L; cognitive dysfunction is reported at levels above 30 mg/L.22,144,146 No active metabolites have been identified.143 Both HPLC and immunoassays are available.
Antimicrobials
Aminoglycosides
Therapeutic ranges. Amikacin, gentamicin, and tobramycin are administered intravenously to treat infections of gram-negative bacilli that are resistant to less toxic antibiotics.42 They are bactericidal, and, thus, their efficacy is highly related to peak concentration after an infusion.152 They also exhibit a postantibiotic effect in which bacterial killing continues even after the serum concentration is below the minimum inhibitory concentration (MIC).91 The concentration-dependent killing and postantibiotic effects of the aminoglycosides explain why extended-interval (pulse) dosing of the aminoglycosides is shown to be safe and effective in many patients. Nephrotoxicity and ototoxicity are the most frequently reported adverse effects of the aminoglycoside antibiotics. Ototoxicity seems to be associated with a prolonged course of treatment (for greater than 7–10 days) with peaks above 12–14 mg/L for gentamicin and tobramycin and 35–40 mg/L for amikacin.91 Patients with trough levels above 2–3 mg/L (gentamicin, and tobramycin) or 10 mg/L (amikacin) for sustained periods of time are predisposed to increased risk of nephrotoxicity.91
Therapeutic ranges for peaks and troughs are reported for the aminoglycosides and pertain only to dosing approaches that involve multiple doses during the day. For gentamicin and tobramycin, peaks between 6 and 10 mg/L and troughs between 0.5 and 2 mg/L are recommended.153 The approximately fourfold higher MIC for amikacin explains why peaks between 20 and 30 mg/L and troughs between 1 and 8 mg/L are recommended.153 There is no therapeutic range when the pulse-dosing method is used; doses are given to attain peaks that are approximately 10 times the MIC, and troughs are intended to be nondetectable within 4 hours of administration of the next dose.152,153 A serum level drawn sometime after infusion of the dose is used only for adjustment of the dosing interval, not to check for efficacy or toxicity.
There has been some concern over the years that aminoglycosides are overmonitored. Uncomplicated patients with normal renal function, who do not have life-threatening infections and will be treated for less than 5 days, may not need to have serum aminoglycoside levels measured.154 At the other extreme, dosage individualization using serum levels of aminoglycosides are absolutely necessary in patients with serious infections who are on prolonged treatment courses, especially if unusual pharmacokinetic parameters are expected (e.g., renal impairment, burns, cystic fibrosis, extremes of age, sepsis, and pregnancy) and if risk of toxicity is high (such as in patients taking concomitant loop diuretics or nephrotoxic drugs [e.g., amphotericin, cyclosporine, or vancomycin]).153,154
Sample timing. For pulse dosing in patients with normal renal function, a steady-state is never reached since each dose is washed out prior to the next dose. The method developed by Nicolau et al. (the so-called Hartford method) requires that a single blood sample be obtained between 6 and 14 hours after the end of the first infusion.155 This sample is referred to as a random sample, but the time of the collection must be documented. The level is used with a nomogram in order to determine if a different dosing interval should be used.42,155 Levels that are too high, according to this nomogram, will indicate that the drug is not being cleared as well as originally predicted, suggesting the need for a longer interval. For traditional dosing, it is important to wait until a steady-state is reached before obtaining blood samples. The half-lives of the aminoglycosides are 1.5–3 hours for adults with normal renal function but as long as 72 hours in patients with severe renal impairment.91 Since the dosing interval for aminoglycosides is usually adjusted to be 2–3 times the drug’s half-life, then a conservative rule of thumb is that steady-state is reached after the third or fourth dose.42 Some patients may have blood samples drawn immediately after the first dose (“off the load”) in order to determine their pharmacokinetic parameters for purposes of dosage regimen individualization. These would most likely be patients who are anticipated to have unpredictable or changing pharmacokinetic parameters, such as those in a critical care unit, and who require immediate effective treatment because of life-threatening infections.
Two blood samples are sufficient for purposes of individualizing traditional aminoglycoside therapy, and will provide reasonable estimates of aminoglycoside pharmacokinetic parameters.156 It is crucial that the times of the sample collections be accurately recorded.42,152 The two samples should be spaced sufficiently apart from each other so that an accurate determination of the log-linear slope can be made in order to determine the elimination rate constant. One sample (sometimes referred to as the measured peak) should be drawn no earlier than 1 hour after the end of a 30-minute, at minimum, infusion.156 A second sample may be drawn any time later but is usually drawn within 30 minutes of the start of infusion of the next dose (assumed to be the trough).42,152,154 If it is expected that the trough level will be close to the limit of the assay sensitivity, the second sample may be drawn earlier.154,156 Once the elimination rate constant has been calculated using these two levels, the true peak and true trough can be calculated and their values compared to desired target peaks and troughs.
Specimens, collection methods, and assays. Serum or EDTA-treated plasma is recommended. Blood collection tubes using gel barriers are acceptable for serum.157,158 Heparin has been shown to interfere with some assays and is not recommended unless the laboratory has ruled out any problems.42 A study showed that gentamicin concentrations in citric acid-stimulated saliva of pediatric patients were good predictors of trough plasma gentamicin concentrations, but only when pulse dosing was used (24-hour dosing interval).159 It was suggested that gentamicin may require a long period of time to fully equilibrate between plasma and saliva, thus explaining the lack of correlation in measured levels when divided doses were used.159 Aminoglycoside concentrations in cerebrospinal fluid are between 10% and 50% of serum concentrations; no therapeutic ranges for cerebrospinal fluid concentrations have been established.42
Serum or plasma should either be assayed within 2 hours of collection, or frozen at 0oC to 5oC.42 This is particularly important for samples that contain beta-lactam antibiotics such as penicillin G, ampicillin, carbenicillin, nafcillin, or ticarcillin.91 The beta-lactam antibiotics, commonly administered with aminoglycosides, physically bind aminoglycoside antibiotics in blood resulting in their inactivation.42,91,152 In vivo, this means the aminoglycoside is cleared more rapidly than usual. The primary concern, however, is continued inactivation of the aminoglycoside that can occur after a blood sample has been collected. A serum concentration that is 7 mg/L at the time of collection might become 6 mg/L after a period of time at room temperature. Use of the artifactually low serum concentration would lead to errors in determination of the aminoglycoside pharmacokinetic parameters. If immediate assay is not possible, the serum or plasma sample should be immediately frozen.
Use of levels for dosage adjustment. Various pulse-dosing methods are used to take advantage of the concentration-related killing and postantibiotic effects of the aminoglycosides.154 The original Hartford method involves giving a mg/kg dose that is administered in order to attain a peak concentration that is approximately 10 times the MIC. Then a sample is obtained between 6 and 14 hours after the end of the infusion and compared to a nomogram, which indicates the appropriate maintenance dosing interval—usually 24, 36, or 48 hours.155 Pulse-dosing methods are not routinely recommended for certain patients, including those with enterococcal endocarditis, renal failure, meningitis, osteomyelitis, or burns.154 However, studies are ongoing to show safety and efficacy in more subpopulations of patients. The results of clinical trials do not consistently show a reduction in nephrotoxicity, and it has been proposed that pulse doses be lowered to provide daily AUC similar to those measured following traditional daily doses.152,160
Serum concentrations of aminoglycosides obtained during traditional dosing are used to determine an individual patient’s pharmacokinetic parameters, as well as the true peak and true trough in order to compare these to desired target levels. Equations that account for time of drug infusion are used to determine an appropriate dosing interval and dose.161 Other dosage adjustment methods include nomograms and population pharmacokinetic (Bayesian) methods.153,161
Protein binding, active metabolites, and other considerations. The aminoglycosides are less than 10% bound to serum proteins, and unbound concentrations will always reflect total concentrations in serum.153 The metabolites of the aminoglycosides are inactive.
Chloramphenicol
Therapeutic range. Chloramphenicol is a broad spectrum antibiotic reserved for treatment of serious infections, including treatment of meningitis caused by ampicillin-resistant Haemophilus influenzae type b.42,162 The therapeutic range for peak levels is generally considered to be 10–20 mg/L. A dose-related reversible type of bone marrow depression may occur and is associated with sustained peak serum levels above 25 mg/L. Irreversible aplastic anemia occurs rarely and is not believed to be related to the serum concentration of chloramphenicol. A somewhat lower therapeutic range may be used for neonates (7.5–14 mg/L) because of the lower serum binding of chloramphenicol in this group—32% versus 53% in adults.99,162 Toxic reactions, including fatalities, have occurred in premature infants and newborns who have had sustained chloramphenicol serum levels above 40–50 mg/L.42,162 These reactions, known as the Gray syndrome, are likely caused by the immature conjugation and renal clearance pathways in these patients.
Chloramphenicol should be monitored closely in patients to guide dosing and avoid toxicity in patients with liver or renal disease or in whom drug–drug interactions are anticipated.42 One study in children, ages 1–66 months, showed progressive decreases in chloramphenicol levels during treatment, suggesting that this group should be frequently monitored.163 It is important that baseline blood counts and hepatic and renal function tests be done before initiation of therapy and repeated during treatment.
Sample timing. The half-life of chloramphenicol is 2–5 hours in adults; steady-state is usually assumed to occur within 12–24 hours.162 The half-life in neonates and infants may range from 8–22 hours.164 Thus, a steady-state should not be assumed in these groups for at least 3 days.
Because both efficacy and toxicity to chloramphenicol are related to peak levels, it is necessary to anticipate when the peak level will occur. Chloramphenicol is available orally as either chloramphenicol base or the chloramphenicol palmitate, which is hydrolyzed to active chloramphenicol in the intestine. Chloramphenicol succinate is the only available intravenous product and is hydrolyzed to chloramphenicol by esterases in the liver, kidneys, and lungs. The peak times for chloramphenicol, therefore, depend not only on the rate of absorption or infusion but also on the rate of hydrolysis in the case of these prodrugs.162 Times associated with peak serum concentrations of chloramphenicol are approximately 1 hour for the orally administered base, 1.5–3 hours for the orally administered palmitate suspension, and between 0.5 and 1 hour after the end of a 30-minute succinate infusion.42 Times of peak chloramphenicol levels following intravenous infusion of the succinate to infants are highly affected by infusion rate, injection site, volume of fluid in the tubing, and type of infusion system.165 It is important that specific guidelines be established at individual institutions to best estimate the times at which peak chloramphenicol levels will occur.
Specimens, collection methods, and assays. Both serum and plasma are acceptable for analysis of chloramphenicol. Gel barrier serum separator tubes have not caused a problem with chloramphenicol.42 There is some suggestion that serum or plasma should be protected from light.42 Also, in vitro hydrolysis of the succinate has been reported, and samples are not stable when stored at –20oC for longer than 1 week.166 The most commonly used assays for chloramphenicol are HPLC and immunoassay (EMIT). The immunoassay method has the necessary sensitivity and specificity but does not permit measurements of palmitate or succinate concentrations.167 High-performance liquid chromatography is sufficiently sensitive and may also permit simultaneous determination of both prodrug and active drug. This ability to determine concentrations of prodrug would be useful only for explaining the reason for a particular chloramphenicol level. For example, low concentrations of chloramphenicol along with high concentrations of the succinate would indicate limited capacity for hydrolysis of the prodrug.162
Cerebrospinal fluid concentrations of chloramphenicol are sometimes measured (in which case the assay must ensure the necessary sensitivity). Concentrations in cerebrospinal fluid need to be above the MIC of the organism, usually between 1 and 6 mg/L.42 Saliva concentrations of chloramphenicol are not reliable predictors of serum chloramphenicol concentrations.168
Use of levels for dosage adjustment. Chloramphenicol has linear elimination characteristics. Therefore, serum chloramphenicol concentrations should change in proportion to the change in daily dose.
Protein binding, active metabolites, and other considerations. Chloramphenicol is 53% to 60% bound in the serum of adults, with lower binding in neonates (32%) and adults with cirrhosis (42%).162 Unbound chloramphenicol concentrations in neonates who have total concentrations between 7.5 and 14 mg/L are similar to unbound concentrations in adults who have total levels between 10 and 20 mg/L.99 None of the metabolites of chloramphenicol show significant activity, and, therefore, do not need to be considered when interpreting chloramphenicol serum concentrations.
Vancomycin
Therapeutic range. Vancomycin, a glycopeptide antibiotic with a narrow spectrum of activity, is used intravenously to treat gram-positive organisms resistant to other antibiotics.42,91 Emergence of vancomycin-resistant enterococci has led to the need to restrict its use. The major toxicities associated with vancomycin are nephrotoxicity and ototoxicity (likely aggravated by concurrent administration of other nephro- and ototoxic drugs). Another adverse effect known as red man syndrome (intense flushing, tachycardia, and hypotension) is usually associated with infusion times shorter than 1 hour.91,169
While many institutions monitor both peaks and troughs of vancomycin, this practice has been questioned, in part because of a lack of standardization of when a sample should be drawn to appropriately reflect a “peak.” In contrast to the aminoglycosides, it is more important to maintain vancomycin levels above the MIC during the dosing interval (to ensure efficacy) than it is to have high peaks and low troughs. It is usually assumed that trough concentrations for vancomycin should be between 5 and 15 mg/L, and greater than 10 mg/L for deep seated infections such as endocarditis.169,170 A range of 15–20 mg/L for troughs is recommended for treatment of pneumonia or other nafcillin- or methicillin-resistant Staphylococcus aureus infections.169,170 While it is sometimes recommended that peak concentrations must be kept below 50 mg/L to avoid ototoxicity, this recommendation is based on only two cases.152 It is more likely that ototoxicity is the result of all levels being too high during the dosing interval (an excessively high total vancomycin exposure).152 Some pharmacokinetic dosing methods are, therefore, based on targeting peak vancomycin serum concentrations (those drawn 2 hours after the end of the infusion) between 30 and 50 mg/L. 169
Vancomycin is routinely monitored in all patients in some hospitals, but many question the need for this in uncomplicated patients with normal renal function.42,152 Indications for monitoring include decreased or changing renal function, especially in patients receiving other nephrotoxic or ototoxic drugs; patients expected to have unusual pharmacokinetics (burns, malignancies, and intravenous drug abusers); patients on therapy for longer than 10 days; patients showing poor response; and patients with unusually high MICs.42,170,171
Sample timing. The half-life of vancomycin is 7–9 hours in adults with normal renal function but can be as long as 120–140 hours in patients with renal failure. Vancomycin half-life is approximately 7 hours in full-term neonates, 6 hours in children, and 12 hours in patients older than 65.170 Half-lives are 3–4 hours in obese patients and 4 hours in burn patients.91 Samples should be obtained as troughs, within 0.5–1 hour of the start of the next infusion.
Specimens, collection methods, and assays. Serum or plasma, using EDTA-treated tubes, may be used. Heparinized tubes should be avoided based on reports of instability of vancomycin in the presence of heparin. There are no reports or problems using serum separator tubes.42
Immunoassays (EMIT and FPIA) are the most common assays used for routine measurements of serum or plasma vancomycin concentration measurements. High-performance liquid chromatography or radioimmunoassay may also be used.170 Serum vancomycin concentrations in patients with renal failure were overestimated by one FPIA method because of cross-reactivity of the polyclonal antibody with a vancomycin crystalline degradation product which accumulates in renal failure patients.172 This same method underestimated serum vancomycin concentrations in patients with hyperbilirubinemia.173 An EMIT method and a modified FPIA method, which both used monoclonal antibodies, did not significantly overestimate vancomycin serum concentrations in these patients.174
Use of levels for dosage adjustment. Vancomycin elimination is linear, and an increase in the dose (without a change in the dosing interval) can be expected to provide a proportional change in the trough serum concentration. It must be cautioned that vancomycin has a very pronounced distribution phase, making the standardization of any so-called peak sample to be especially important. More sophisticated prediction methods for dosing adjustments must be used if the dosing interval is adjusted with or without a change in dose.
Many methods have been proposed for vancomycin dosage regimen adjustment.91,169,175,176 A relatively simple method proposed by Ambrose and Winter permits the use of a single trough level (drawn within 1 hour of the start of the next infusion) along with an assumption of the population distribution volume to predict the necessary pharmacokinetic parameters needed for individualization.169 Once those parameters are determined, the aminoglycoside individualization equations can be used to target desired peak and trough vancomycin concentrations.
Protein binding, active metabolites, and other considerations. Vancomycin is 30% to 55% bound to serum proteins in adults with normal renal function. The binding is lower (19%) in patients with end-stage renal disease.176 With binding this low, total concentrations of vancomycin will always provide reliable indications of the unbound concentrations in serum. Vancomycin metabolites are inactive, and, thus, do not contribute to antibacterial effect or toxicity.
Amphotericin B
While amphotericin B continues to be considered the drug of choice for most systemic fungal infections, serum concentration monitoring is not recommended.177 The nephrotoxic effects of amphotericin B do not appear to be related to serum concentration, and the range of concentrations associated with beneficial effect is, likewise, unclear.177
Flucytosine
Therapeutic range. Flucytosine is a synthetic antifungal agent that is often used in combination with amphotericin B for treatment of systemic fungal infections.178 It is also used increasingly in combination with the azole antifungal agents and is part of a new therapeutic approach in the treatment of certain tumors, such as colorectal carcinoma.179 Most clinicians agree that peak serum concentrations of flucytosine should be kept below 100 mg/L to avoid dose-related hepatotoxicity, bone marrow depression, and gastrointestinal disturbances.152,179,180 Some clinicians also advise that trough concentrations of flucytosine be kept between 25 and 50 mg/L (or kept above 25 mg/L) in order to avoid rapid development of resistance.152,178,179 If a constant infusion is used, steady-state serum concentrations of 50 mg/L should be targeted.152 The hepatotoxicity and bone marrow suppression are both usually reversible with discontinuation. Indications for monitoring flucytosine include avoidance of toxicity—particularly in patients with impaired renal function or those receiving concomitant amphotericin B—and avoidance of resistance due to sustained low levels.152,178–180
Sample timing. The half-life of flucytosine is approximately 3–4 hours in patients with normal renal function; it is usually advised to wait 24 hours before a steady-state is assumed.179,180 The half-life can be as long as 85 hours in patients with renal failure in which case steady-state would not be reached for approximately 10 days.178 Peak concentrations should be obtained 1–2 hours after an oral dose or 30 minutes after the end of an infusion.152,177–179 The peak time occurs later after an oral dose of flucytosine in patients with poor renal function because of either slowed absorption or a shift in peak time related to the drug’s longer half-life.179 Trough concentrations, if indicated, should be drawn within 30 minutes of the next dose.
Specimens, collection methods, and assays. Serum is the most common specimen reported for analysis. There do not appear to be special precautions for blood collection devices. The most common assays include microbiological, GLC, HPLC, and an automated enzymatic method.178,180,181 The enzymatic method compares well to HPLC but shows some degree of nonspecificity with icteric and lipemic samples.181
Use of levels for dosage adjustment. Because there are no reports of nonlinear elimination behavior, a given increase in dose rate or infusion rate should produce a proportional increase in serum flucytosine concentration.
Protein binding, active metabolites, and other considerations. Flucytosine does not exist as enantiomers, has no active metabolites, and is minimally bound to serum proteins.
Azole Antifungals
Therapeutic ranges. While serum concentrations of the azoles have been measured and documented following successful therapy, serum concentrations associated with toxicity have not been clearly documented. Serum concentrations of ketoconazole between 1.5 and 6 mg/L, and of fluconazole between 30 and 90 mg/L have been associated with effective chronic therapy.177,180 Efficacy has been associated with the following serum concentration ranges of the triazole antifungals, as measured by HPLC methods: 0.5–2 mg/L for itraconazole, 0.5–1.5 mg/L for posaconazole, and 0.5–2 mg/L for voriconazole.182,183
The primary reason for monitoring the azole antifungal drugs is to ensure efficacy. Itraconazole levels are known to be relatively low in patients with AIDS or acute leukemia, most likely due to malabsorption and concurrent administration of enzyme-inducing drugs.184 For this reason, some consider the serum concentration monitoring of itraconazole to be essential in patients with life-threatening fungal infections.177 Ketoconazole is recommended for monitoring only in patients with treatment failure or relapse, or if drug–drug interactions or malabsorption are suspected.177,180 Fluconazole is the least likely to require monitoring, as its absorption is predictable and it is less affected by drug–drug interactions.177
Sample timing. The half-lives of fluconazole, itraconazole, and posaconazole range between 24 and 31 hours.182 Thus, steady-state will not be attained for at least 1 week after initiation of therapy or adjustment of the dosage regimen. The half-lives of ketoconazole and voriconazole are shorter (3–6 hours) and steady-state can therefore be expected after 24–48 hours.180,182 Since the purpose of monitoring the azoles is to ensure that minimum levels of drug are present, trough levels should be obtained when possible.
Specimens, collection methods, and assays. Serum is the most common specimen reported for analysis of the azole drugs. There do not appear to be any reported problems associated with blood collection methods. The most common assays for the azoles include microbiological (bioassay), GLC, and HPLC.177,180,184 Because itraconazole has an active metabolite that may be present at concentrations that are 2–3 times higher than the parent drug, concentration readings using microbiologic assays will be higher than those reported using the chromatographic methods.183,184 An HPLC method that measures both the parent and hydroxylated metabolite is preferred for itraconazole. Although concentrations of fluconazole in stimulated saliva were highly correlated with concentrations in plasma, the accuracy of plasma concentration prediction was not adequate.185
Use of levels for dosage adjustment. Although azole levels are not used for the purpose of dosage adjustment, fluconazole, ketoconazole, fluconazole, and posaconazole exhibit first-order elimination behavior, and increases in dose rate or infusion rate can be expected to produce proportional increases in drug concentrations.177 Itraconazole and voriconazole are reported to have nonlinear elimination behavior, such that greater-than-proportional increases in serum drug concentration should be expected with increases in dose rate.152,183
Protein binding, active metabolites, and other considerations. Itraconazole, ketoconazole, and posaconazole are 98% to 99% bound to serum proteins, primarily albumin, For these drugs, it is possible that some of the inability to correlate total concentrations with response and toxicity is complicated by variable serum protein binding among patients. Voriconazole is 60% bound, while fluconazole is only 12% bound.183 Itraconazole concentrations in the presence of variable quantities of the active metabolite may also complicate the correlation of itraconazole serum concentrations with effect and toxicity.183
Antimycobacterials
The optimal use of therapeutic drug monitoring for mycobacterial infections is currently under study. Drugs that are FDA-approved and considered first line as part of an initial four-drug regimen are isoniazid, rifampin, pyrazinamide, and either ethambutol or streptomycin. Of these, isoniazid and rifampin are the most important based on their relatively high potency and favorable side-effect profiles. Second-line agents that are more toxic must be used if drug resistance emerges and include ethionamide, cycloserine, capreomycin, para-aminosalicylic acid, and dapsone.186
It is essential that adequate levels of these antimycobacterial drugs be present in serum for effective treatment. This does not always occur, even in patients in whom adherence has been documented.186 Lower-than-expected levels of antimycobacterial drugs have been reported in patients with diabetes and in those with HIV infections, which in some cases was associated with malabsorption.187-189 There is also considerable potential for drug–drug interactions among the antimycobacterial drugs, given the effects of rifampin, isoniazid, and the fluoroquinolones in either inducing or inhibiting cytochrome P450 isozymes.190 Drugs used to treat HIV patients may also contribute to this drug–drug interaction quagmire.
A study in non-HIV infected tuberculosis patients who were not responding to treatment as expected showed that 29% to 68% of them had serum antimycobacterial drug levels below target ranges.191 In another study, a small percentage of nonresponding patients all showed suboptimal levels of rifampin.192 After dosage adjustments were made, all patients responded to treatment. The authors recommended that low serum rifampin levels be suspected in patients who do not respond after 3 months of supervised drug administration, or earlier in patients with HIV infection, malnutrition, known gastrointestinal or malabsorptive disease, or hepatic or renal disease.
Specialized laboratories have been developed that offer sensitive and specific assays for serum concentrations for the most commonly used antimycobacterial drugs.186 As more specific information about the efficacy of therapeutic drug monitoring of these drugs becomes available, more laboratories and services of this type will likely be available.193
Antiretrovirals
Therapeutic ranges. There is some evidence that favors limited serum concentration monitoring of drugs used in the treatment of HIV-1 infection, in particular the PIs and the nonnucleoside reverse-transcriptase inhibitors (NNRTIs).194,195 These drugs, particularly the PIs, show marked interpatient variability in their pharmacokinetics, and retrospective studies show strong relationships between drug concentrations and virologic response.196 In addition, suboptimal levels of the antiretroviral drugs are associated with acquired drug resistance and virologic failure.197 A substudy of the randomized, prospective clinical trial, ATHENA, showed that patients who underwent serum drug concentration monitoring for the antiretroviral drugs had a significantly higher likelihood of virological response as compared to those who did not undergo monitoring.196
Minimum effective concentrations have been determined for the most common PIs based on in vitro determinations of drug concentrations (corrected for serum binding) required for 50% or 90% inhibition of replication in the patient’s virus isolate (IC50 or IC90). Attention has turned more recently, however, to the use of a new parameter that may be a better predictor of response. The inhibitory quotient (IQ) is the ratio of the patient’s trough plasma concentration to the IC50 or IC90.196 A high IQ would indicate more drug is present in the patient than is needed for virologic response, while a low IQ would indicate inadequate drug levels or a resistant virus. Recent studies show virologic response may be better related to IQ than to trough levels alone.196 Future studies may focus on the definition of therapeutic ranges of IQ rather than minimum concentrations.
The most commonly used PIs are fosamprenavir (a prodrug of amprenavir), darunavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir and tipranavir. The most commonly used NNRTIs are efavirenz, nevirapine and etravirine. Serum concentrations of newer agents (enfuvirtide, a fusion inhibitor, and raltegravir, an integrase inhibitor) may also be monitored in select situations.194,195 Some clinicians advocate the monitoring of these drugs in all patients on initiation of therapy to ensure adequate levels; others reserve use for selected situations including patients with renal or liver disease, pregnancy, children, patients at risk for drug interactions, and suspected toxicity.196,198
Sample timing. Half-lives of the NNRTIs average 25–50 hours, and steady-state will be reached after a week in most patients.199 However, a steady-state will be reached within 2 days for the PIs, which have half-lives ranging from 2–12 hours.199 Predose samples are recommended as the minimum effective concentrations and the IQs are based on the lowest drug concentration during the dosing interval. There may be logistic problems with this timing, however, in cases when the drug is administered once daily in the evening. Some drugs, such as nelfinavir, exhibit a lag in their absorption, such that the lowest concentration actually occurs about an hour after administration of the next dose.
Specimen, collection methods, and assays. Serum or plasma has been used as the specimen for analysis. It is important that the possible influence of gel barrier or serum separator tubes be determined by the laboratory prior to use. Given the high binding of these drugs to AAG, tubes with stoppers possibly formulated with TBEP must be avoided. Nevirapine concentrations in citric-acid simulated saliva strongly correlated with concentrations in plasma and plasma ultrafiltrate.200 Indinavir concentrations in saliva also show promise as noninvasive alternatives to plasma concentrations.201 Concentration monitoring in saliva for the other antiretroviral drugs is not likely to be as promising because they are highly protein bound (>90%), and assay sensitivity would be limiting.
High-performance liquid chromatography is most commonly used to determine serum concentrations of the antiretroviral drugs, and HPLC methods have been developed to measure as many as seven PIs and two NNRTIs in a single run using ultraviolet absorption.202,203
Use of levels for dosage adjustment. Dosage adjustments of the antiretroviral drugs, for the most part, should result in proportional changes in the trough serum drug concentration, provided the dosing interval is not altered. Reports showing serum drug concentrations to be unpredictable after dosage adjustments in some patients suggest that nonadherence with antiretroviral regimens is a major concern.196 Serum concentrations of amprenavir, lopinavir, nelfinavir, and saquinavir may be difficult to maintain above their minimum effective concentrations because of rapid clearances and large first-pass effects. Rather than increasing their dose rate, ritonavir, a potent inhibitor of CYP3A4-mediated metabolism in the gut wall and liver, may be coadministered as a pharmacoenhancer. This results in decreased gastrointestinal enzyme metabolism of the PI, higher trough levels, and, in most cases, prolonged elimination half-lives.204
Protein binding, active metabolites, and other considerations. The serum protein binding of nevirapine and indinavir is 50% to 60%, while the protein binding of the other antiretrovirals is greater than 90%.196,199 Alpha-1-acid glycoprotein and albumin are the primary binding proteins for these drugs in serum.196 As would be expected, there is considerable variability in the unbound fraction of these drugs in serum. In addition, AAG concentrations are elevated in patients with HIV-1 infection and can return to normal with treatment. Thus, the same total level of the drug would be expected to reflect a lower level of response early in treatment as compared to later. Clearly, total concentrations of the PIs and NNRTIs should be cautiously interpreted if unusual serum binding is anticipated, but no clear guidelines are yet available. Only nelfinavir has a metabolite that is known to be active.196 While studies indicate the measurement of the metabolite is probably not crucial, there is likely to be considerable variability among and within patients in the presence of this metabolite.
Cardiac Drugs
Digoxin
Therapeutic ranges. There has been a dramatic reduction in digoxin toxicity since the advent of therapeutic drug monitoring for digoxin.62 Digoxin’s inotropic effect is the basis for its use for treatment of congestive heart failure, while its chronotropic effects are the basis for treatment of atrial arrhythmias such as atrial fibrillation, atrial flutter, and paroxysmal atrial tachycardia. The commonly reported therapeutic range is 0.5–2.0 mcg/L in adults and 1–2.6 mcg/L in neonates.62,99,205 The lower end of the range (0.5–1 mcg/L) is generally used for treatment of heart failure, with levels up to 1.5 mcg/L possibly leading to additional benefit.81 Higher serum digoxin concentrations are required for treatment of atrial arrhythmias (0.8–1.5 mcg/L), with additional benefit gained in some patients with levels up to 2 mcg/L.91
Fifty percent of patients with serum digoxin concentrations above 2.5 mcg/L show some form of digoxin toxicity.91 Symptoms of toxicity include muscle weakness; gastrointestinal complaints (anorexia, nausea, vomiting, abdominal pain, and constipation); CNS effects (headache, insomnia, confusion, vertigo, and changes in color vision); and serious cardiovascular effects (second- or third-degree atrioventricular [AV] bradycardia, premature ventricular contractions, and ventricular tachycardia).91,205 In fact, many of the cardiac arrhythmias observed with high digoxin concentrations resemble the clinical condition being treated, hence the need to monitor serum digoxin concentrations to distinguish toxicity from inadequate therapy.
There are several physiologic or pathologic conditions that can shift the therapeutic range of digoxin. Its toxicity may be more likely within the therapeutic range if the patient has hypokalemia, hypomagnesemia, hypercalcemia, or underlying heart disease (e.g., coronary atherosclerotic heart disease or an old myocardial infarction).25,62 Patients with hyperthyroidism are believed to be more resistant to digoxin.25
The primary indications for digoxin monitoring include (1) suspected digoxin toxicity in order to determine the appropriate amount of antidote (digoxin-immune Fab fragments; Digibind®) needed; (2) suspected poisoning from ingestion of plants or herbal medications that contain structurally similar glycosides; (3) impaired renal function to adjust the dose rate; and (4) suspected interactions with drugs such as antacids, amiodarone, oral antibiotics, cholestyramine, cyclosporine kaolin-pectin, metoclopramide, neomycin, quinidine, spironolactone, sulfasalazine, verapamil, and St. John’s wort.25,206
Sample timing. The average digoxin half-life in adults with normal renal function is approximately 2 days; at least 7 days are recommended to attain a steady-state.205 In the case of treatment of digoxin overdose with digoxin-immune Fab fragments (a fragment of an antibody that is very specific for digoxin), blood samples for serum digoxin measurements should not be obtained sooner than 10 days after administration of the fragments.62,205 Since most immunoassays measure both the free and Fab-bound digoxin, premature sampling would lead to artifactually high digoxin concentration readings.
Samples drawn during the absorption and distribution phases after administration of digoxin cannot be appropriately interpreted by comparison to the usual therapeutic range. Digoxin levels in blood do not reflect the more important levels in myocardial tissue until at least 6 hours after the dose (some say at least 12 hours).205-207 Blood samples should, therefore, be drawn anytime between 6 hours after the dose and right before the next dose (Figure 5-5).
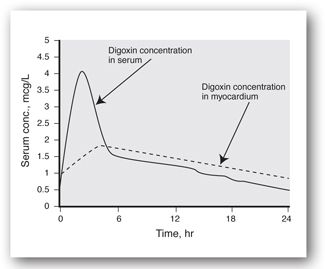
FIGURE 5-5. Simulated plot showing concentrations of digoxin in serum (mcg/L) and concentrations in myocardial tissue (units not provided) after a dose of digoxin at steady-state. Tissue concentrations do not parallel concentrations in serum until at least 6 hours after the dose.
Inappropriate timing of samples for digoxin determinations is a major problem in hospitals. One study showed that 55% of the samples submitted to the laboratory for digoxin analysis lacked clinical value because of inappropriate timing.208 In another study, standardization of digoxin administration times for 1700 and blood sample times for 0700 resulted in a dramatic reduction in inappropriately timed samples.209 Another recommendation is that the laboratory immediately contact the clinician if digoxin levels are above 3.5 mcg/L.205 If it is confirmed that the sample was drawn too early after the dose, another sample should be requested. Alternatively, the laboratory should collect sample timing information as part of the laboratory request form and refuse to assay any samples that are inappropriately timed.
Specimens, collection methods, and assays. Serum or plasma, anticoagulated with heparin or EDTA, may be used. In general, serum separator tubes should be avoided.205 Serum is recommended if ultrafiltration is to be done for purposes of determining unbound concentrations of digoxin in patients treated with Fab fragments (Digibind®). Samples are stable for 24 hours at 2oC to 8oC and 1–2 weeks at –20oC.205 Saliva concentrations of digoxin have been measured in a number of studies, but none show a sufficiently strong correlation with either total or unbound levels in serum. Part of the poor correlation was proposed to be related to active secretion of digoxin into saliva or interferences from endogenous digoxin-like immunoreactive substances (DLIS).159
Digoxin serum concentrations are measured almost exclusively using commercial immunoassay methods.62,205 The digoxin antibodies used in these immunoassays cross-react to varying extents with digoxin metabolites, endogenous DLIS, and other drugs and their metabolites (spironolactone and its active metabolite canrenone, digitoxin, and digitoxin metabolites). In fact, immunoassays for digoxin may cross-react with structurally similar substances in Chinese medicines (e.g., dried venom of Chinese toad) or plants (oleander) and, therefore, may be used to detect the presence of these substances, which cause digoxin-like toxicity.205 It is important to note that these interferences can result in overreading or underreading of digoxin. One study comparing nine different commercial immunoassay methods showed that three of the nine methods underreported potentially toxic digoxin concentrations because of negative interferences caused by spironolactone and canrenone.71 Potentially interfering digoxin metabolites will accumulate in renal impairment; DLIS are predominant in the blood of patients with renal and liver disease, those who are pregnant, and in neonates.62 The high potential for interferences reinforces the importance of monitoring signs and symptoms in addition to serum levels.
One major cause of interference with digoxin serum concentration determinations by immunoassay is the presence of Fab fragments (Digibind®) used as an antidote to digoxin toxicity. The digoxin antibodies from the immunoassay cause this interference by competing with the Fab fragments for digoxin in the same samples. In patients with renal failure, this source of interference persists for more than 10 days after administration of the antidote. Ultrafiltration of the serum sample removes the digoxin-bound Fab fragments and permits a fairly reliable measurement of the unbound digoxin concentration.210 Newer assays have been developed to directly measure unbound digoxin in the presence of Digibind® without ultrafiltration.211
Use of levels for dosage adjustment. Dose rate adjustments of digoxin based on serum digoxin concentrations are straightforward. Because of linear elimination behavior, a given increase in the daily digoxin dose will produce a proportional increase in the serum concentration at that time during the dosing interval. Again, it is extremely important that a serum level used for dose rate adjustment be obtained no earlier than 6–8 hours after the last dose.
Protein binding, active metabolites, and other considerations. Digoxin is only 20% to 30% bound to serum proteins.25 Therefore, total concentrations in serum will reflect the pharmacologically active unbound concentration. The biologic activity of digoxin metabolites is modest compared to the parent drug, and variable presence of metabolites should not affect the interpretation of a digoxin serum concentration.
Lidocaine
Therapeutic range. Lidocaine is a Type 1B antiarrhythmic used as second-line therapy for the acute treatment for ventricular tachycardia and fibrillation. The therapeutic range is generally considered to be 1.5–5 mg/L with concentrations greater than 6 mg/L considered to be toxic.91,205,212 Minor side effects—drowsiness, dizziness, euphoria, and paresthesias—may be observed at serum concentrations above 3 mg/L. More serious side effects observed at concentrations above 6 mg/L include muscle twitching, confusion, agitation, and psychoses, while cardiovascular depression, AV block, hypotension, seizures, and coma may be observed at concentrations above 8 mg/L.91,205,213
Lidocaine is not monitored as commonly as some of the other cardiac drugs because its effect (abolishment of the ECG-monitored arrhythmia) is easy to directly observe. Indications should be restricted to situations in which the expected response is not evident (inefficacy or toxicity) or when decreased hepatic clearance is suspected or anticipated: liver disease, congestive heart failure, advanced age, severe trauma, and/or concurrent drugs such as beta-adrenergic blockers, fluvoxamine, or cimetidine.62,205,212
Sample timing. The half-life of lidocaine ranges from 1.5 hours to as long as 5 hours in patients with liver disease.91,213 Thus, steady-state may not be attained for 18–24 hours even if a loading dose is administered. Because lidocaine is administered as a continuous infusion, there are no fluctuations in levels, and blood for lidocaine serum concentration determinations can be drawn anytime at steady-state.
Specimens, collection methods, and assays. Blood collected in serum separator tubes and tubes using TBEP-containing rubber stoppers have resulted in artifactually low lidocaine levels. A new formulation of the Becton-Dickinson serum separator tube, SST II®, was shown to be acceptable, however, with complete recovery of lidocaine from serum stored for as long as 7 days.44 Plasma is also acceptable as a specimen if heparin or EDTA is used as the anticoagulant.175 Lidocaine is stable in serum or plasma stored 24 hours at 2oC to 8oC or 1–2 weeks at –20oC.175
Immunoassays for determination of lidocaine are commercially available and show little cross-reactivity with lidocaine metabolites.62 They do not permit separate determination of the primary active metabolite, monoethylglycinexylidide (MEGX), which has 80% to 90% of the activity of lidocaine.91,205 Chromatographic methods (HPLC and GLC) are preferred if monitoring of the active metabolite is deemed necessary.
Use of levels for dosage adjustment. Adjustments of lidocaine infusion rate should result in a proportional increase in lidocaine serum concentration.
Protein binding, active metabolites, and other considerations. The unbound percentage of lidocaine is normally 30% but can range from 10% to 40% due to variations in AAG concentrations.91,205 The AAG concentrations are decreased in patients with nephrotic syndrome and increased in conditions of trauma, after surgery, and in patients with rheumatoid arthritis, cancer, and morbid obesity.205 Thus, higher total concentrations may be considered therapeutic for patients with higher AAG concentrations.
The AAG concentrations are also increased after a myocardial infarction, resulting in a lower unbound lidocaine fraction during prolonged infusions of lidocaine in these patients.214 The combination of higher total levels of lidocaine during prolonged infusions (believed to be due to competition between lidocaine and its accumulated metabolites) and a lower unbound fraction mean that unbound lidocaine concentrations during prolonged infusions are probably therapeutic.215 It is important to be aware that total lidocaine levels at the higher end of the therapeutic range may not present a danger of toxicity in patients receiving prolonged infusions of lidocaine after a myocardial infarction.
The MEGX metabolite of lidocaine has 80% to 90% of the antiarrhythmic potency of lidocaine, and its concentration accumulates in renal failure. Thus, MEGX may contribute to the pharmacologic effects of lidocaine in patients with renal impairment.91,212
Procainamide
Therapeutic range. Procainamide is not used much anymore in the oral form, but may be used intravenously for select indications, such as patients with atrial fibrillation or flutter who require acute conversion to normal sinus rhythm.216 The therapeutic range of procainamide is complicated by the presence of an active metabolite, NAPA, which has different electrophysiologic properties. Procainamide is a Type 1A antiarrhythmic, while NAPA is a Type III antiarrhythmic.62,91,205 The enzyme that acetylates procainamide is bimodally distributed, such that patients are either slow or fast acetylators. In addition, NAPA is more dependent on the kidneys for elimination than is procainamide, and its levels accumulate more than procainamide for a given level of renal impairment.205,214 Thus, the ratio of NAPA to procainamide in serum can be quite variable, necessitating the use of separate therapeutic ranges for the parent and metabolite.
Most patients respond when serum procainamide concentrations are between 4 and 8 mg/L; some receive additional benefit with levels up to 12 mg/L.216 There have been reports of patients requiring levels between 15 and 20 mg/L without adverse effects.216 Serum concentrations of NAPA associated with efficacy are reported to be as low as 5 mg/L and as high as 30 mg/L. Most clinicians consider toxic NAPA levels to be above 30–40 mg/L.205 Some clinicians feel that NAPA does not need to be monitored except in patients with renal impairment.216 Most laboratories, however, automatically measure both procainamide and NAPA concentrations in the same sample. The practice of summing the two concentrations and comparing to a therapeutic range for summed procainamide and NAPA (often reported as 10–30 mg/L) is to be discouraged.91,205,216 To do this validly, the molar units of the two chemicals would need to be used.62 More importantly, however, is the fact that procainamide and NAPA have completely different electrophysiologic behaviors. The best practice is to independently compare each chemical to its own reference range.62,91,205
Side effects to procainamide and NAPA are similar. Anorexia, nausea, vomiting, diarrhea, weakness, and hypotension may be seen with procainamide levels above 8 mg/L, while levels above 12 mg/L may be associated with more serious adverse effects: heart block, ventricular conduction disturbances, new ventricular arrhythmias, and even cardiac arrest.91 A syndrome known as torsades de pointes may also be seen after procainamide administration, although this is more commonly seen after quinidine or disopyramide administration.91
Indications for procainamide and NAPA serum level monitoring include recurrence of arrhythmias that were previously controlled, suspected toxicity or overdose, anticipated pharmacokinetic alterations caused by drug–drug interactions (including amiodarone, cimetidine, ethanol, ofloxacin, quinidine, ranitidine, and trimethoprim), and disease state changes (renal failure or congestive heart failure, in particular).91,205,214,216
Sample timing. The half-life of procainamide in adults without renal impairment or congestive heart failure ranges from 2.5 hours (fast acetylator) to 5 hours (slow acetylator).91,205 The half-life of NAPA is longer, averaging 6 hours in patients with normal renal function, and 30 hours or longer in patients with renal impairment.91,216 Thus, a steady-state of both chemicals is not observed until at least 18 hours in patients with good renal function or as long as 4 days in renal impairment.
Specimens, collection methods, and assays. Serum or plasma, anticoagulated with heparin, EDTA, or oxalate, may be used.205 Recovery of procainamide and NAPA are not affected by use of serum separator tubes, but the influence of any special blood collection devices should always be confirmed by individual laboratories.205 Serum and plasma are stable for 24 hours at 2oC to 8oC, and for 1–2 weeks at –20oC.205 Saliva levels of procainamide and NAPA have been shown to correlate strongly with plasma levels and are proposed as acceptable, noninvasive alternatives to blood sampling.217
The most commonly used commercial, automated assays for procainamide and NAPA are FPIA and EMIT. These methods require separate determinations of procainamide and NAPA on the same serum sample. Samples that are hemolyzed, lipemic, or icteric may affect the reading of these immunoassay methods.205 Chromatographic methods (HPLC and GLC) allow the simultaneous measurement of procainamide and NAPA and are not subject to interferences from hemoglobin, lipids, or bilirubin.205
Use of levels for dosage adjustment. The 24% lower clearance of procainamide at higher dose rates has been attributed to nonlinear hepatic clearance.218 The clinician should be aware that increases in infusion rate may produce somewhat greater-than-proportional increases in serum procainamide concentration in some patients, particularly those with serum levels at the upper end of the therapeutic range.
Protein binding, active metabolites, and other considerations. Procainamide is only 10% to 20% bound to serum proteins.62,205 Thus, total procainamide and NAPA levels always reflect the pharmacologically active unbound concentrations of these drugs.
Quinidine
Therapeutic range. The therapeutic range of quinidine for treatment of severe malaria due to P. falciparum is reported as 3–8 mg/L.219 When used in combination with verapamil for prevention of atrial fibrillation, the therapeutic range of quinidine is reported to be 2–6 mg/L.220 Common side effects are gastrointestinal in nature (anorexia, nausea, and diarrhea) and more serious side effects include cinchonism (blurred vision, lightheadedness, tremor, giddiness, and tinnitus), hypotension, and ventricular arrhythmias.91,205 Torsades de pointes is more likely to occur at concentrations at the lower end of the therapeutic range, thus complicating the interpretation of quinidine concentrations.62
Indications for monitoring of quinidine concentrations include therapeutic confirmation; suspected toxicity; recurrence of arrhythmias after initial suppression; suspected drug–drug interactions or other conditions known to alter quinidine pharmacokinetics; suspected nonadherence; and changes in administered formulation.91,205,220
Sample timing. The half-life of quinidine is reported to range from 4–8 hours in adults and up to 10 hours in patients with liver disease. Steady-state should be attained within 2 or 3 days, and most clinicians agree that samples should be drawn as a trough within 1 hour of the next dose.62,91,205,220
Specimens, collection methods, and assays. Serum or plasma may be used. Plasma should be collected in EDTA-treated tubes. Serum separator tubes should generally be avoided.205 Quinidine in serum or plasma is stable for 1–2 weeks at –20oC.205
Quinidine in serum or plasma is most frequently assayed using immunoassay, but HPLC may also be used. Dihydroquinidine (an impurity in quinidine dosage forms), quinine, and the quinidine metabolite, 3-hydroxyquinidine, may all interfere to varying extents with immunoassay methods.205 Moderate (20%) cross-reactivity with the 3-hydroxy metabolite was reported with an FPIA method.62
Use of levels for dosage adjustment. Quinidine displays linear elimination behavior for most patients; a change in daily quinidine dose will cause a proportional change in the average steady-state serum quinidine concentration. Nonlinear elimination may be evident in some patients, due either to saturable first-pass metabolism or saturable renal tubular secretion.220 Thus, a greater-than-proportional increase in average quinidine concentration with increase in daily quinidine dose may be evident in some patients.
Protein binding, active metabolites, and other considerations. Quinidine is a weak base that is normally between 70% and 80% bound to albumin and AAG in the serum of healthy patients.205 These protein levels are known to increase in trauma, myocardial infarction, cardiac surgery, atrial fibrillation/flutter, and congestive heart failure, and the percentage binding of quinidine can increase to as high as 92% in these patients.91 The unbound fraction of quinidine was shown to be decreased in patients with atrial fibrillation or atrial flutter, and the unbound quinidine concentration was shown to correlate better with ECG interval changes than total quinidine.221,222 All of this suggests that total serum concentrations of quinidine must be cautiously interpreted in patients with suspected elevations in AAG concentrations. A total quinidine level that is above 5 mg/L could be therapeutic with respect to unbound quinidine concentration.
The dihydroquinidine impurity may be present in amounts that are between 10% and 15% of the labeled amount of quinidine and is believed to have similar electrophysiologic properties as quinidine.205 The 3-hydroxyquinidine metabolite has activity that is less than the parent (anywhere between 20% and 80% have been reported) and is not as highly bound to serum proteins.214 Although not reported, this leads one to wonder about possible accumulation of these substances in renal failure patients with a resultant shift in the quinidine therapeutic range.
Other Cardiac Drugs
Amiodarone. Amiodarone is used for the treatment of life-threatening recurrent ventricular arrhythmias that do not respond to adequate doses of other antiarrhythmics. The primary metabolite, desethylamidarone, has similar electrophysiologic properties as amiodarone and accumulates to levels similar to or higher than the parent drug, especially in renal failure patients.62 The concentration versus effect relationship for amiodarone is poorly defined; some say that serum concentrations between 0.5 and 2.5 mg/L are associated with effectiveness with minimal toxicity.62 The occurrence of toxicity, however, appears to be more reliably related to the total amount of drug administered rather than serum concentration. Laboratories that measure serum amiodarone concentrations report only the parent drug, despite high levels of the active metabolite. In general, therapeutic drug monitoring of amiodarone is of limited benefit because activity of the drug is mostly associated with concentrations in the tissue.62 Serum concentrations might be most useful in cases of suspected nonadherence.
Disopyramide. Disopyramide is used to treat life-threatening ventricular arrhythmias in selected patients.205 Disopyramide has several characteristics that confound the use of serum disopyramide concentration monitoring. It is administered as a racemic mixture, and only the S(+) enantiomer is believed to significantly contribute to the drug’s antiarrhythmic effect.26 Both enantiomers demonstrate concentration-dependent binding, such that increases in dose rate produce proportional increases in unbound (pharmacologically active) enantiomer but less-than-proportional increases in total summed enantiomer concentration.26,62,205 The primary metabolite of disopyramide, mono-N-dealkyldisopyramide, has 50% of the antiarrhythmic activity of the parent but 2–4 times the anticholinergic activity, which is responsible for many of the side effects.205 The metabolite accumulates more than disopyramide in renal failure patients. Despite all of these confounding factors, most laboratories monitor summed enantiomer levels of total parent drug only (no metabolite) and, in most cases, rely on a therapeutic range between 2 and 5 mg/L with levels greater than 7 mg/L considered toxic.205 The use of serum disopyramide concentrations as a guide to dosage adjustments is, understandably, on the decline. Indications that might be appropriate include suspected toxicity or nonadherence and drug–drug interactions or diseases that are anticipated to affect the pharmacokinetics of the enantiomers.205
Flecainide. Flecainide is used for prevention of paroxysmal atrial fibrillation/flutter or paroxysmal supraventricular tachycardias.205 It is administered as a racemic mixture, but unlike disopyramide, there is little difference in the pharmacologic effects of these enantiomers.62 The commonly used therapeutic range for trough concentrations, based on the flecainide acetate salt, is 0.2–1 mg/L; the range based on the flecainide base is 0.175–0.870 mg/L.62,205 Toxicity is likely observed at acetate concentrations greater than 1.6 mg/L.62 Although this is a fairly wide therapeutic range, the pharmacokinetics of flecainide are quite variable among patients, suggesting that serum concentration monitoring might be helpful. Indications for monitoring may include patients who have a recent myocardial infarction, impaired renal function, or in whom drug–drug interactions are suspected.205 The serum binding of flecainide is low (32% to 58%) so that total flecainide concentrations provide a reliable reflection of the active, unbound concentration. Flecainide is assayed by FPIA or HPLC.205
Mexiletine. Mexiletine is structurally similar to lidocaine but has the advantage that it can be given orally. It is used for the treatment of life-threatening ventricular arrhythmias in select patients and may also be used for treatment of chronic pain syndromes.62 It is given as a racemic mixture—with the S(+) enantiomer showing greater activity than the R(–) enantiomer—and is only 50% to 60% bound to serum proteins. Mexiletine is usually assayed by achiral methods (GLC or HPLC); no studies have yet been done to relate effect to individual enantiomers.62 The therapeutic range for the summed total enantiomers is most commonly reported to be 0.5–2.0 mg/L.62,214 Toxicity may occur, however, at concentrations within the range of effective concentrations.214 Mild side effects may be seen between 0.8 and 3 mg/L, and severe side effects between 1 and 4.4 mg/L.214 Because the extent of mexiletine absorption can be significantly affected by changes in the rate of gastric emptying, patients receiving narcotics and those who have had a recent myocardial infarction might benefit from serum concentration monitoring.214 Higher serum levels of mexiletine may be seen in patients with liver disease and patients with congestive heart failure.223
Cytotoxic Drugs
While cytotoxic drugs have some characteristics that make them ideal candidates for therapeutic drug monitoring (narrow therapeutic indices and variable pharmacokinetics) they have many more characteristics that make therapeutic drug monitoring difficult or unsuitable.224,225 They lack a simple, immediate indication of pharmacologic effect in order to aid definition of a therapeutic range (the ultimate outcome of cure could be years). They are given in combination with other cytotoxic drugs, such that concentration versus effect relationships for any single drug is difficult to isolate. They are used to treat cancer, which is a highly heterogeneous group of diseases, each possibly having its own concentration versus effect relationships. Finally, many of these drugs require tedious assay techniques. In summary, cytotoxic drugs are not routinely monitored because they are in need of more clearly defined therapeutic ranges. If ranges are established, they are usually more helpful to avoid toxicity than to define zones for efficacy.
Methotrexate
Therapeutic range. Methotrexate is the only antimetabolite drug for which serum concentrations are routinely monitored.224 It acts by blocking the conversion of intracellular folate to reduced folate cofactors necessary for cell replication. While cancer cells are more susceptible to the toxic effects of methotrexate, healthy host cells are also affected by prolonged exposure to methotrexate. It is for this reason that leucovorin, a folate analogue that prevents further cell damage, is administered following high-dose methotrexate treatments.224 Measurements of serum methotrexate concentrations at critical times following high-dose methotrexate regimens are imperative to guide the amount and duration of leucovorin rescue treatments, thus preventing methotrexate toxicity. Institution of protocols for methotrexate serum concentration monitoring for this purpose has resulted in dramatic reductions in high-dose methotrexate-related toxicity and mortality.226
While it is known that methotrexate levels must be sufficiently high in order to prevent relapse of the malignancy, the specific range of levels defining efficacy has been difficult to define.226 However, the relationship between methotrexate levels and toxicity has been much more clearly defined. Depending on the protocol, methotrexate levels that remain above 0.1 µM for longer than 48 hours are associated with a high risk of cytotoxicity. 227 Prolonged levels of methotrexate can lead to nephrotoxicity, myelosuppression, gastrointestinal mucositis, and liver cirrhosis.224,226
Serum concentration monitoring is not generally indicated when relatively low doses of methotrexate are given for chronic diseases such as rheumatoid arthritis, asthma, psoriasis, and maintenance for certain cancers.
Sample timing. The timing of samples for determination of methotrexate concentrations is highly dependent on the administration schedule. As one example of such a protocol, a methotrexate dose may be administered by intravenous infusion over 36 hours followed by a regimen of leucovorin doses administered over the next 72 hours.227 Additional or larger leucovorin doses might be given depending on the methotrexate levels in samples drawn at various times after the start of the methotrexate infusion. It is important that methotrexate levels continue to be monitored until they are below the critical levels (usually between 0.05 and 0.1 µM).224,227
Specimen, collection methods, and assays. Serum or plasma concentrations are generally used. Saliva concentrations correlate poorly with total and unbound methotrexate concentrations, precluding the clinical use of saliva as a noninvasive alternative for blood samples.228 Rapid reporting of methotrexate levels is important, and the immunoassay methods are preferred for determination of methotrexate levels. All immunoassays have different specificities and sensitivity limits and none stand out as clearly superior.226 The most widely used method, FPIA, offers a sensitivity limit as low as 0.02 µM, and minimal cross-reactivity with the major metabolite, 7-hydroxymethotrexate.226 Chromatographic methods such as HPLC must be used if quantitation of methotrexate metabolites is desired, most likely for research purposes.
Use of levels for dosage adjustment. Methotrexate and leucovorin doses are based on protocols.
Protein binding, active metabolites, and other considerations. Methotrexate binds to albumin in serum ranges from 20% to 57%.224 While studies have shown the unbound fraction of methotrexate to be increased by concomitant administration of NSAIDs, salicylate, sulfonamides, and probenecid, the implications for interpretation of methotrexate levels are probably not important.227 The methotrexate metabolite, 7-hydroxymethotrexate, has only 1/100th the activity of methotrexate but may cause nephrotoxicity due to precipitation in the renal tubules.226
Petros and Evans provide an excellent summary of cytotoxic drugs and the types of measurements that have been used to predict their toxicity and/or response.226 Correlations between response or toxicity and serum concentration or area under the serum concentration (AUC) versus time curve for total drug have been shown for bisacetamide, busulfan, carboplatin, cisplatin, cyclophosphamide, docetaxel, etoposide, 5-fluorouracil hexamethylene, irinotecan, paclitaxel, teniposide, topotecan, and vincristine.226,229,230 The strong correlation between busulfan AUC and bone marrow transplant outcome led the FDA to include instructions for AUC monitoring in the package insert for intravenous busulfan. Unbound AUC values for etoposide and teniposide, which demonstrate concentration-dependent binding, correlate more strongly with toxicity than corresponding total plasma AUC values.231 Systemic drug clearance has been predictive of response/toxicity for amsacrine, fluorouracil, methotrexate, and teniposide.226,232 Steady-state average serum concentrations or concentrations at designated postdose times have also been predictive of response/toxicity for cisplatin, etoposide, and methotrexate.226 Finally, concentrations of cytosine-arabinoside metabolite in leukemic blasts and concentrations of mercaptopurine metabolite in red blood cells have been predictive of response or toxicity for these drugs. Correlations between systemic exposure and response/toxicity for cyclophosphamide, carmustine, and thiotepa have also been reported.233
Other than methotrexate, none of the assays for these drugs (often done by HPLC) is available commercially as an immunoassay.226 While most studies up to this point have focused on use of cytotoxic drug concentration measurements to minimize toxicity, future studies will be increasingly focused on use of drug concentrations to maximize efficacy.
Immunosuppressants
Cyclosporine
Therapeutic range. Cyclosporine is a potent cyclic polypeptide used for prevention of organ rejection in patients who have received kidney, liver, or heart transplants. It is also used for the management of psoriasis, rheumatoid arthritis, and other autoimmune diseases. The therapeutic range of cyclosporine is highly dependent on the specimen (whole blood or serum/plasma) and assay. Most transplant centers use whole blood with one of the more specific assays—HPLC or immunoassays that use monoclonal antibodies (monoclonal radioimmunoassay or monoclonal fluorescence polarization immunoassay).91,234,235 The commonly cited therapeutic range for whole blood troughs using one of these specific methods is 100–500 mcg/L.91,236 Troughs at the higher end of this range may be desired initially after transplantation and in patients at high risk for rejection.236 Therapeutic ranges are lower if serum or plasma is used and higher if a less specific assay is used, such as an immunoassay based on polyclonal antibodies. The therapeutic range also depends on the specific organ transplantation procedure and the stage of treatment after surgery (higher concentrations during induction and lower concentrations during maintenance to minimize side effects).61,91,235-237 Thus, it is important that the therapeutic range guidelines established by each center be used. While most centers still use single trough levels to adjust cyclosporine doses, the area under the blood concentration versus time curve is believed to be a more sensitive predictor of clinical outcome.238 Studies that have investigated the use of single cyclosporine concentrations measured 2 hours after the dose, as a surrogate for the AUC value, suggest a better clinical outcome as compared to the use of single trough levels.234,238,239
Cyclosporine has a narrow therapeutic index and extremely variable pharmacokinetics among and within patients. The implications of ineffective therapy and adverse reactions are serious. Thus, it is imperative that cyclosporine concentrations be monitored in all patients starting immediately after transplant surgery. The primary side effects associated with high cyclosporine blood concentrations are nephrotoxicity, neurotoxicity, hypertension, hyperlipidemia, hirsutism, and gingival hyperplasia.61,91,236 Blood cyclosporine concentrations should also be monitored when there is a dosage adjustment, signs of rejection or adverse reactions, suspected nonadherence, or initiation or discontinuation of drugs known to induce or inhibit cyclosporine metabolism.61,236
Sample timing. Monitoring is often done immediately after surgery before a steady-state is reached. Initially, levels may be obtained daily or every other day, then every 3–5 days, then monthly. Changes in dose rate or initiation or discontinuation of potential enzyme inducers or inhibitors will require resampling once a new steady-state is reached. The half-life of cyclosporine ranges from 5–27 hours and is dependent on the particular formulation. Thus, 3–5 days is generally adequate in most patients for attainment of a new steady-state. Most centers continue to sample predose (trough) cyclosporine levels, while some are using 2-hour postdose levels, which appear to more closely predict total exposure to cyclosporine as measured by AUC.234,239 Multiple samples to determine the AUC are generally unnecessary.
Specimens, collection methods, and assays. Blood concentrations of cyclosporine are 2–5 times the concentration in serum because of extensive partitioning into red blood cells. Whole blood is the preferred specimen and should be collected in tubes with EDTA.61 Samples are stable for 7 days in plastic tubes at 4oC.240 Capillary blood by skin puncture is also acceptable.61 While the popular monoclonal immunoassays offer improved specificity over the older polyclonal versions, they continue to measure varying amounts of cross-reactive metabolites.61,234 The reference method of HPLC offers optimal specificity but takes longer; it might be considered in selected cases where significant interferences from metabolites are suspected.
Use of levels for dosage adjustment. In most cases, simple proportionality may be used for dosage adjustments. Trough or 2-hour postdose levels will change in proportion to the change in dose rate so long as the dosing interval remains the same.
Protein binding, active metabolites, and other considerations. Cyclosporine is 90% bound to albumin and lipoproteins in blood.237 Unbound fractions in blood vary widely among patients and are weakly correlated to lipoprotein concentrations in blood.237 For example, lower unbound fractions of cyclosporine have been reported in patients with hypercholesterolemia.61 Lindholm and Henricsson reported a significant drop in the unbound fraction of cyclosporine in plasma immediately prior to acute rejection episodes.241 An association between low cholesterol levels (and presumably high unbound fractions of cyclosporine) and increased incidence of neurotoxicity has also been reported.61 These studies suggest that efforts to maintain all patients within a certain range of total concentrations may be misleading. Routine monitoring of unbound cyclosporine levels is not yet feasible, given the many technical difficulties with this measurement. Instead, the clinician should cautiously interpret total levels of cyclosporine in situations where altered protein binding of cyclosporine has been reported.
Other Immunosuppressants
Tacrolimus. Tacrolimus is a macrolide antibiotic with immunosuppressant activity and is generally used in combination with other immunosuppressant drugs. Like cyclosporine, whole blood is the preferred specimen.242 Trough blood concentrations of tacrolimus as high as 20 mcg/L are targeted during initial treatment and gradually decrease to between 5 and 10 mcg/L during maintenance therapy, often after 12 months.55 Toxicities to tacrolimus are very similar to those with cyclosporine, including nephrotoxicity and neurotoxicity.236,242 The unpredictable and variable extent of tacrolimus bioavailability (5% to 67%) contribute to the need for monitoring of this drug.242 Monitoring should always be done after changes in dose rate or initiation/discontinuation of enzyme-inducing or inhibiting agents. The half-life of tacrolimus ranges from 4–41 hours, and a new steady-state will be attained after approximately 3–5 days.61,236 While trough concentrations are still the method of choice for monitoring, a second level might be needed if Bayesian approaches to dosage individualization are used.239,243 The majority of centers use immunoassay methods, either a microparticle enzyme immunoassay (MEIA) or EMIT, which show some cross-reactivity with tacrolimus metabolites. More specific methods, such as HPLC, may be required in patients with liver disease who are anticipated to have high levels of metabolites.61 Bilirubin and alkaline phosphatase do not interfere with measurements of tacrolimus using MEIA, but abnormally high hematocrit levels have caused underreading of tacrolimus.244 Tacrolimus appears to exhibit linear elimination behavior. Thus, an increase in the daily dose is expected to result in a proportional increase in the steady-state trough level. Tacrolimus is 75% to 99% bound to plasma proteins (albumin, alpha-1-glycoprotein, lipoproteins and globulins).61,242 Reports of lower unbound serum concentrations of tacrolimus during episodes of rejection lead one to be cautious with interpretation of total tacrolimus concentrations in patients with suspected alterations in protein binding.245
Mycophenolic acid. Mycophenolate mofetil, the prodrug of mycophenolic acid, is often used in combination with cyclosporine or tacrolimus with or without corticosteroids.61 While troughs of mycophenolic acid may be monitored (plasma levels between 2.5 and 4 mg/L are targeted with good success), AUC values appear to be better predictors of postoperative efficacy (avoidance of acute rejection).239,246,247 Reliable measurements of AUC may be determined with as few as three samples (trough, 30 minutes, and 120 minutes postdose) with a desired target AUC range of 30–60 mg × hr/L.246 The half-life of mycophenolic acid is approximately 17 hours. Thus, a new steady-state will be attained approximately 3 days after a dose rate change or the addition/discontinuation of drugs that affect the metabolism of mycophenolic acid. In contrast to cyclosporine, tacrolimus, and sirolimus, plasma anticoagulated with EDTA is the preferred specimen for mycophenolic acid concentration measurements.61,246 The acyl glucuronide metabolite of mycophenolic acid is active, but its clinical significance for the interpretation of mycophenolic acid levels is not yet clear. This metabolite cross-reacts with immunoassay using the EMIT method, giving higher readings as compared to HPLC.61 Mycophenolic acid is 98% bound to plasma proteins, and the unbound fraction is greatly influenced by changes in albumin concentration, displacement by metabolites, renal failure, and hyperbilirubinemia.234,246 Several groups of investigators suggest that unbound mycophenolic acid concentrations should be monitored when altered binding is suspected.61,246-249 There is evidence that unbound mycophenolic acid concentration may be a better predictor of adverse effects.245,248
Sirolimus. Sirolimus is a macrolide antibiotic with potent immunosuppressant activity. When used in combination with cyclosporine and corticosteroids, trough blood concentrations of 5–15 mcg/L are generally targeted.236 It has a relatively long half-life (62 hours), and a new steady-state will not be attained in many patients until at least 6 days after dose rate adjustments or the addition/discontinuation of interacting drugs.236 At present, trough concentrations are used for monitoring as they correlate well with AUC.234 Whole blood is the preferred specimen and should be collected using EDTA as the anticoagulant.250 Samples are not stable at temperatures above 35oC but may be stored at room temperature for up to 24 hours, between 2oC and 8oC for up to 7 days, and at 20oC for up to 3 months.250 An MEIA method is under development, but it overestimates sirolimus concentrations measured by HPLC because of cross-reactivities with sirolimus metabolites.61
Psychotropics
Amitriptyline, Nortriptyline, Imipramine, Desipramine
Therapeutic ranges. Although tricyclic antidepressants (TCAs) continue to be used for treatment of depression, their use has significantly declined in favor of the newer antidepressants which have more favorable side effect profiles.251 The therapeutic ranges of amitriptyline, imipramine, desipramine, and nortriptyline are well-defined.21,252,253 Desipramine and nortriptyline, while drugs in their own right, are also active metabolites of imipramine and amitriptyline, respectively. Thus, amitriptyline is included as a fourth TCA for serum concentration monitoring.
When imipramine is administered, combined serum concentrations of imipramine and desipramine that are considered therapeutic but not toxic are between 180 and 350 mcg/L.252,253 Combined levels above 500 mcg/L are extremely toxic.253 When desipramine is administered, levels between 115 and 250 mcg/L are frequently associated with therapeutic effect.252,253 When amitriptyline is administered, combined serum concentrations of amitriptyline and nortriptyline should be between 120 and 250 mcg/L.252 Combined levels above 450 mcg/L are not likely to produce additional response and are associated with cardiotoxicity anticholinergic delirium.253 The therapeutic range of nortriptyline is the most firmly established of these four drugs; target serum concentrations after nortriptyline administration are between 50 and 150 mcg/L.252-254
The most common side effects of the TCAs are anticholinergic in nature—dry mouth, constipation, urinary retention, and blurred vision.252 Toxicities seen at higher concentrations include cardiac conduction disturbances (with prolonged QRS interval evident on EKG), seizures, and coma.252 For all of the TCAs, these toxic effects occur at serum concentrations that are approximately 5 times those needed for antidepressant efficacy.12
Since it takes 3 or more weeks beyond the final dose rate adjustment to fully assess clinical response to a TCA, routine monitoring once a steady-state has been attained would shorten the overall dosage titration period as compared to a trial-and-error dose rate adjustment method. A dosage individualization method, using serum nortriptyline levels drawn following the first dose, resulted in patients being discharged 6 days earlier and returning to work 55 days earlier as compared to a control group.37 Response rates to TCAs are reported to increase from 30% to 40% to as high as 80% by use of serum TCA concentration monitoring.255 Other indications for monitoring include suspected nonadherence or inadequate response, suspected toxicity, and suspected unusual or altered pharmacokinetics (children, elderly, and drug interactions).
Sample timing. While the half-lives of amitriptyline and nortriptyline range from 9–56 hours with a steady-state attained within 4–11 days in most patients, the half-lives of imipramine and desipramine range from 6–28 hours with a steady-state attained in 6 days.253,254 As a general rule, the clinician should wait at least a week before drawing any blood samples for serum concentration monitoring. Trough levels are ideal for monitoring purposes because they are the most reproducible. Troughs are inconvenient, however, since patients usually take the drug once daily at bedtime. Therefore, a standardized sampling time is commonly used—12 to 14 hours after the bedtime dose. 253,254 Since this sample is taken midway through the dosing interval, it provides a fairly good approximation of the average steady-state level of TCA. If the TCA happens to be given in divided doses, a 4- to 6-hour postdose sample time is recommended.252,254
Specimens, collection methods, and assays. Serum or plasma collected using EDTA as the anticoagulant is preferred. There is some suggestion that heparin lowers the concentrations of TCAs, and there were numerous reports in the literature about TCAs having spuriously low serum levels due to displacement from AAG by TBEP in the rubber stopper.252 Although the stopper has since been reformulated, it is a good idea to avoid blood collection materials that have not been tested by the laboratory, as well as special serum separator tubes with gel barriers.252 Serum or plasma should be immediately separated from red blood cells to avoid the possibility of hemolysis.252 Serum or plasma can be stored for 24 hours at room temperature, for 4 weeks at 4oC, or for more than 1 year at –20oC.
Chromatographic and immunoassay methods are most commonly used for measurements of serum TCA concentrations with immunoassay being most common.253,254 Of the two available immunoassay methods, only the EMIT system uses a monoclonal antibody to determine individual concentrations of these four TCAs. An FPIA method measures for the presence of all tricyclic drugs (total tricyclics) using a less specific polyclonal antibody and is, therefore, useful for toxicology screenings.252 False positives may result from such screenings, however, if carbamazepine is present.256 One limitation of the EMIT method is that the tertiary amines (imipramine, amitriptyline, and others) cross-react with one another, while the secondary amines (desipramine and nortriptyline) also cross-react with one another.252 This becomes a problem if the patient is receiving more than one TCA or is being switched from one to another.
Use of levels for dosage adjustment. Some mild nonlinearity has been described for desipramine, in which case, increases in the daily dose will be expected to produce somewhat greater-than-proportional increases in the standardized 12-hour sample. The other TCAs, however, exhibit proportionality. A method proposed by Browne et al. for the dosing of nortriptyline involves the use of a serum level drawn following a first dose to predict an appropriate maintenance dosage regimen.37 This method, very similar to methods used for initiation of lithium therapy, is not commonly used, but has demonstrated great promise in speeding up the dose-titration period.
Protein binding, active metabolites, and other considerations. The TCAs in general are highly bound to serum proteins, including albumin, AAG, and lipoproteins.251 Based on this, one would expect unbound TCA serum concentrations to be much better predictors of response than total concentrations, particularly in populations suspected to have unusually high or low serum binding. Until now, studies that have attempted to examine this have not been able to clarify relationships between response and total serum concentrations based on variable protein binding. Assays for accurate and direct measurement of unbound TCA concentrations in serum need to be sufficiently sensitive for these kinds of studies.
The TCAs are extensively metabolized and undergo significant first-pass metabolism. While the primary active metabolites have been identified and are separately measured, other active metabolites can accumulate in some circumstances and affect the response at a given parent drug concentration. In one study, levels of conjugated and unconjugated hydroxylated metabolites of the TCAs were markedly elevated in patients with renal failure and believed to contribute to the hypersensitivity of these patients to TCA side effects.257
Lithium
Therapeutic range. Lithium is a monovalent cation used for the treatment of bipolar disorder and the manic phase of affective disorders. The concentration units for lithium are expressed as mEq/L, which is the same as mmol/L. While the overall therapeutic range for treatment of manic depression is commonly cited as 0.5–1.2 mEq/L.21 there appear to be two distinct ranges used in practice, depending on the stage of therapy.252 For acute management of manic depressive episodes, the therapeutic range of 0.5–1.2 mEq/L is desired, going up to 1.5 mEq/L if necessary.91,258,259 For maintenance treatment, the therapeutic range of 0.6–0.8 mEq/L is usually recommended.91,258,259 Serum concentrations above 1.5 mEq/L are associated with fine tremors of the extremities, gastrointestinal disturbances, muscle weakness, fatigue, polyuria, and polydipsia. Concentrations above 2.5 mEq/L are associated with coarse tremors, confusion, delirium, slurred speech, and vomiting. Concentrations above 2.5–3.5 mEq/L are associated with seizures, coma, and death.252 It is important to point out that the values for the therapeutic ranges are based on samples obtained at a specific time during the day—just before the morning dose and at least 12 hours after the evening dose for patients on a BID or TID regimen.259,260
Most clinicians require that every patient taking lithium be regularly monitored, which is cost-effective considering the potential avoidance of toxicity.91,254 Specific indications for lithium concentration monitoring include evaluation of nonadherence; suspicion of toxicity; confirmation of the level associated with efficacy; and any situation in which altered pharmacokinetics of the drug is anticipated (drug–drug interactions, pregnancy, children, geriatric patients, and fluid and electrolyte imbalance). Despite the strong indication for lithium monitoring in all patients, 37% of lithium users on Medicaid did not have serum drug concentrations monitored.261
Sample timing. The half-life of lithium ranges from 18–24 hours, and steady-state will be reached within a week of therapy.259 However, 2–3 weeks of treatment may be required after that before the full response to the drug can be assessed.259 When initiating lithium therapy, it is recommended that serum levels be measured every 2–3 days (before a steady-state is reached) to ensure that levels do not exceed 1.2 mEq/L during that time.91 Because of the extreme variability of serum lithium levels during the absorption and distribution periods, the current standard of practice is to draw all samples for lithium serum concentration determination 12 hours after the evening dose, regardless of whether a twice or thrice daily dosing schedule is used. For example, the time for blood sampling for a patient on a 0900/1500/2100 schedule would be right before the 0900 dose.91 The timing of blood samples for a patient taking once daily lithium is less clear, given the greater degree of serum lithium concentration fluctuation with this dosing method.259
Specimens, collection methods, and assays. While plasma is acceptable, it must be collected using sodium heparin as the anticoagulant, not lithium heparin. Serum is the preferred specimen, therefore, just to avoid any possible confusion.215 The serum sample should be rejected if there is any evidence of hemolysis since release of high concentrations of lithium from the red blood cells will artifactually raise the serum concentration.252 To minimize the chance of hemolysis, serum should be separated from the red blood cells within 1 hour of collection.254 Other than one exception, involving an interference from a silica clot activator, blood collection tubes generally have not introduced any artifacts for lithium assays.262 Lithium in serum or plasma is stable at room temperature or refrigeration temperature for extended periods of time.252
Lithium erythrocyte concentration has been proposed to correlate better with response and toxicity since it represents intracellular lithium.252 This method, however, has never been routinely adopted for monitoring. Saliva concentrations of lithium have been proposed as noninvasive substitutes for serum or plasma lithium concentrations. However, the S:P ratio is quite variable, even within some patients. The average S:P ratio ranges from two to four and is affected by many variables. If the S:P ratio is shown to be stable for an individual patient over time, saliva monitoring might prove to be useful for some patients.263 Based on an audit of clinical laboratories in Europe and the United Kingdom, flame emission photometry and atomic absorption spectroscopy are still the most commonly used methods for lithium quantitation and offer excellent precision, accuracy, and few interferences.264 Ion-selective electrode methods are more rapid and less costly but may have problems with interferences. Carbamazepine, quinidine, procainamide, NAPA, lidocaine, and valproic acid can all produce biases in lithium measurements by ISE.252 High calcium levels may also produce a positive bias with some ISE methods.252 A new colorimetric dry slide-based serum lithium assay is concluded to offer an acceptable alternative to currently available methods for monitoring lithium.265
Use of levels for dosage adjustment. Lithium exhibits linear elimination behavior and proportionality can be assumed when dosage adjustments are made. The assumption of linearity is the basis for several dosing methods that are used for initiating lithium therapy in patients. The Cooper method involves drawing a sample for lithium analysis 24 hours after a first dose of 600 mg.38 The resulting level, believed to provide a reflection of the drug’s half-life, is used with a nomogram that indicates the optimal maintenance regimen. The Perry method requires that two levels be drawn during the postabsorption-postdistribution phase after a first dose of lithium.39 These two levels are used to determine the first-order elimination rate constant, which can then be used to determine the expected extent of lithium accumulation in the patient. The maintenance regimen required to attain a desired target lithium concentration in that patient can then be determined. Population-pharmacokinetic dosing-initiation methods (Bayesian) can also be used.91
Protein binding, active metabolites, and other considerations. Lithium is not bound to serum proteins, nor is it metabolized.
Other antidepressants. Assays have been developed to document the serum concentrations observed following administration of other cyclic antidepressants as well as the selective serotonin reuptake inhibitors, serotonin and norepinephrine reuptake inhibitors, and norepinephrine reuptake inhibitors.12,253,258,266-268 While reference ranges have been established, there does not appear to be any compelling reason for routine monitoring of these drugs given their relatively wide therapeutic indices and more favorable side effect profiles. Because 50% of patients do not achieve optimal relief from symptoms of depression, some clinicians advocate the use of serum concentration monitoring in patients who do not initially respond to identify nonadherence or to identify unusually low serum concentrations.12,21,252,253
Antipsychotics. The existence of well-defined therapeutic ranges for most antipsychotic drugs remains controversial.255,269,270 There is some justification, however, for serum concentration monitoring of clozapine, fluphenazine, haloperidol, olanzapine, perphenazine, risperidone, and thioridazine in special circumstances.21,270,271 Reference ranges for other antipsychotic drugs are primarily based on average serum concentrations observed during chronic therapy.254,255,272 One difficulty in establishing clear therapeutic range guidelines is that chronicity of illness and duration of antipsychotic drug exposure can shift the therapeutic range; separate therapeutic ranges may need to be developed depending on duration of illness.272
FUTURE OF TDM
Drug assays are rapidly improving with regard to specificity, sensitivity, speed, and convenience. Methods that separate drug enantiomers may help to elucidate therapeutic ranges for compounds administered as racemic mixtures.273 Capillary electrophoresis-based assays will be increasingly used in clinical laboratories because of their low cost, specificity, utility for small sample volumes, and speed.274 Methods for measurement of drugs in hair samples are being proposed for assessment of long-term drug adherence.275 Implanted amperometric biosensors, currently used for glucose monitoring, may be useful for continuous monitoring of drug concentrations.276 Subcutaneous microdialysis probes may also be useful for continuous drug monitoring, particularly since they monitor pharmacologically active unbound drug concentrations.277 Point-of-care assay methods, currently used in private physician offices, group practices, clinics, and emergency rooms, could eventually be used in community pharmacies in the future.278,279
The therapeutic drug monitoring of the near future may also involve determination of genotypes, characterization of proteins produced in particular diseases (proteomics), and analysis of drug metabolite profiles (metabonomics).280,281 These sciences may help to identify those subsets of patients who will be nonresponders or toxic responders, and help to determine appropriate initial doses. Such testing would not require special sample timing, might be possible using noninvasive methods (e.g., hair, saliva, and buccal swabs), and would need to be done only once as the results would apply over a lifetime. These types of testing may help patients to receive the best drug for the indication and rapid individualization of drug dosage to achieve desired target concentrations.28,29,280,282 This will likely result in increased demand for new types of tests from clinical laboratories currently involved in routine therapeutic drug monitoring.
There is a movement to change the terminology and practice of therapeutic drug monitoring to target concentration strategy, target concentration intervention, or therapeutic drug management.40,283 Critics of the therapeutic drug monitoring terminology claim that it suggests a passive process that is concerned only with after-the-fact monitoring to ensure that levels are within an ill-defined range without proper regard to evaluation of the response to the drug in an individual patient.283 Target concentration intervention is essentially a new name for a process used by clinical pharmacokinetics services for years and involves the following steps: (1) choosing a target concentration (usually within the commonly accepted therapeutic range) for a patient; (2) initiating therapy to attain that target concentration using best-guess population pharmacokinetic parameters; (3) fully evaluating response at the resulting steady-state concentration; and (4) adjusting the regimen as needed using pharmacokinetic parameters that have been further refined by use of the drug concentration measurement(s).
Methods to improve the therapeutic drug monitoring process itself are needed. Every effort should be made to focus on patients who are most likely to benefit from therapeutic drug monitoring, and minimize time and money spent on monitoring that provides no value.9 The biggest problems with the process continue to be lack of education, communication, and documentation.6,19 Approaches to changing physician behavior with regard to appropriate sampling and interpretation include educational sessions; formation of formal therapeutic drug monitoring services; multidisciplinary quality improvement efforts; and computerization of requests for drug concentration measurement samples.284 Pharmacists will continue to have a pivotal role in the education of physicians and others involved in the therapeutic drug monitoring process. Future studies that evaluate the effect of therapeutic drug monitoring on patient outcomes will likely use quality management approaches.285
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


