CHAPTER 34 Inherited disorders of coagulation
Introduction
This chapter will focus on the inherited disorders of coagulation, ranging from the more common disorders such as von Willebrand disease and hemophilia A and B to a number of the so-called ‘rare congenital bleeding disorders’. Inherited and acquired disorders affecting platelets, acquired hemorrhagic disorders and the thrombophilias will be discussed elsewhere (Chapters 32, 33, 35 and 36 respectively).
Hemophilia
Inheritance
Hemophilia A and B are X-linked recessive disorders and thus affect males almost exclusively. Mothers and daughters of affected males are, by definition, obligate carriers. Rarely a female may manifest symptomatic hemophilia through one of the following mechanisms: 1) homozygous female offspring of an affected male and a carrier female; 2) high degree of lyonization (skewed X-chromosome inactivation) of alleles in a carrier; and 3) hemizygosity of females with concomitant Turner syndrome.1 Females who appear phenotypically hemophilic should also undergo evaluation to exclude von Willebrand disease and testicular feminization syndrome.
The molecular basis of hemophilia A and B (FVIII and FIX genes)
Hemophilia A is more common than hemophilia B possibly because the factor VIII (FVIII) gene is considerably larger and thereby more susceptible to spontaneous mutation. The cloning of the FVIII gene and the sequencing of the cDNA was reported in landmark papers in 1984.2–4 The FVIII gene comprises around 186 000 base pairs, compared to the FIX gene which has approximately 34 000 base pairs. In fact, the FVIII gene is one of the larger genes in the human genome, accounting for about 0.1% of the X chromosome. It contains three identifiable domain types in the sequence A1-A2-B-A3-C1-C2. This sequence comprises a heavy chain (A1 and A2 domains), a connecting region (B domain) and a light chain (A3, C1, and C2 domains). Some of these domains have specific functions, such as binding to factor IXa (A2 domain with A1/A3-C1-C2 dimer) while different epitopes of the C2 domain bind to phosphatidylserine (a procoagulant phospholipid expressed on the surface of activated platelets and endothelium) as well as von Willebrand factor, thrombin, and factor Xa. The B-domain is not required for procoagulant activity. The FVIII gene possesses 26 exons; within intron 22 are the start points for two further genes, one entirely contained within the intron and apparently expressed in most tissues (F8A) and a second beginning within the intron and utilizing exons 23–26 of the FVIII gene itself (F8B).5
After the FVIII gene was cloned, it became apparent that there was not a uniform genotypic abnormality that accounted for all cases of hemophilia A, and a variety of responsible mutations has now been described. The reader is referred to an online resource for the known mutations of factor VIII known as HAMSTeRS (Hemophilia A Mutation, Structure, Test, and Resource Site), which can be found at http://hadb.org.uk (see also Table 34.1). The most common mutation resulting in severe hemophilia A involves one of several inversions within intron 22 that collectively account for approximately 45–50% of cases. These mutations result in failure of transcription across this intron due to inversion of a section of the X chromosome at the tip of the long arm, resulting in the separation of the factor gene into two parts (Fig. 34.1). Recognition of this mutation has had a significant impact on carrier detection, as it is usually the first and most rapidly identifiable mutation sought.6 Failure to identify an intron 22 inversion in severe hemophilia is an indication to evaluate for a much less common defect in intron 1 (present in <5% of cases) and then complete gene sequencing. In contrast to severe hemophilia A, moderate and mild hemophilia A are usually due to missense mutations in the FVIII gene.6
Table 34.1 Available online resources for documented mutations in coagulation factor deficiency states
| Fibrinogen | www.hgmd.org www.geht.org/databaseang/fibrinogen/ |
| Prothrombin | www.coagMDB.org/ |
| Factor V | An up-to-date database on FV mutations has been complied by Dr. Hans L. Vos and is available upon request (email: H.L.Vos@lumc.nl) |
| Factor VII | www.coagMDB.org/ |
| Factor VIII | http://hadb.org.uk |
| Factor IX | www.kcl.ac.uk/ip/petergreen/haemBdatabase.html; www.coagMDB.org/ |
| Factor X | www.coagMDB.org/ |
| Factor XI | www.FactorXI.org/ |
| FXIII | www.f13-database.de/(xhgmobrswxgori45zk5jre45)/index.aspx |
| VWF | www.ragtimedesign.com/vwf/mutation www.vwf.group.shef.ac.uk/ |
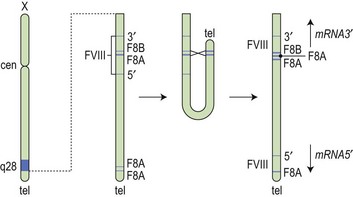
Fig. 34.1 How the tip flips: the mechanism of inversion through intron 22. cen, centromere; tel, telomere.
(Reproduced with permission from Hoffbrand AV, Mitchell Lewis S, Tuddenham EGD, eds. Postgraduate Haematology. Butterworth Heinemann, Oxford, 1999).
The FIX point mutations and smaller deletions typically result in production of a nonfunctioning but immunologically detectable, FIX protein (‘cross-reacting material positive’ or CRM+). Hemophilia resulting from large to complete deletions or nonsense mutations is more likely to be CRM−. Patients that are CRM− are more susceptible to the development of FIX alloantibodies, which overall are relatively uncommon in hemophilia B compared to hemophilia A.7 The Hemophilia B Mutation Database can be found online at http://www.kcl.ac.uk/ip/petergreen/haemBdatabase.html (Table 34.1).
As alluded to above, inhibitory alloantibodies that develop in a proportion of patients with hemophilia A and B following replacement therapy correlate to some extent with factor VIII and factor IX mutation type and location;8 this aspect is discussed in greater detail below.
Incidence and clinical manifestations
The incidence of hemophilia A is estimated at 1 : 5000 live male births (Table 34.2). Factor VIII deficiency accounts for approximately 80% of hemophilia. Hemophilia B is much less common, with an estimated incidence of 1 : 30 000 live male births. Notably, the hemophilias have equal incidence across racial and ethnic groups.
Of all patients with hemophilia, approximately 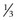 –
–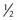 will have severe disease. In general, hemophilia A and B of a corresponding severity manifest a similar clinical picture. It is estimated that approximately 50% of severe and 30% of mild/moderate hemophilia cases are without significant family history and are considered the result of a spontaneous mutation.9
will have severe disease. In general, hemophilia A and B of a corresponding severity manifest a similar clinical picture. It is estimated that approximately 50% of severe and 30% of mild/moderate hemophilia cases are without significant family history and are considered the result of a spontaneous mutation.9
Some patients with severe hemophilia may have a milder clinical course. For example, patients with the Leyden phenotype of hemophilia B have severe disease in childhood that becomes mild after puberty. This is thought to be secondary to a mutation in the promoter region that disrupts the binding site for hepatocyte nuclear factor-4 (HNF-4) but not an overlapping site for an androgen response element. This responsiveness to male sex hormones may explain the milder clinical course that emerges after puberty.10–12
Patients with severe hemophilia may also co-inherit a hereditary prothrombotic condition such as a prothrombin G20210A gene mutation or factor V Leiden that may partially offset their hemophilia and result in fewer or less severe hemorrhagic episodes.13–16
Bleeding may occur in any part of the body, but most commonly affects the joints, followed by muscles and the gastrointestinal tract. The joints most commonly involved are listed in decreasing order of incidence: knees (50% of all bleeding episodes), elbows, ankles, shoulders and wrists.17,18
Severe hemophiliacs may develop a pseudotumor. These are chronically unresolved hematomas produced by repetitive bleeding episodes into muscle that slowly enlarge and become encapsulated and organized over time. The accompanying inflammatory process may eventually encroach or destroy surrounding structures, frequently including bone. Unfortunately pseudotumors are more common in areas of the world where there is often inadequate treatment of hemophilia. Immediate and appropriate treatment of acute bleeding episodes theoretically minimizes the risk of pseudotumor formation. Even with appropriate factor replacement, surgical removal of large pseudotumors is associated with up to a 20% mortality.19
Bleeding into the central nervous system is a particularly ominous complication, and is an all too frequent cause of death.20 In children, particularly in the neonatal period, intracranial hemorrhage (ICH) may occur with minimal or no recognized trauma. In adults, 50% of cases of ICH appear to be spontaneous. HIV infected hemophiliacs treated with protease inhibitors may be at higher risk for spontaneous intramuscular or ICH.21 Of patients who experience an ICH, approximately 50% will develop permanent neurologic sequelae, and up to 30% will die.
Treatment of hemophilia
The care of patients with hemophilia is complicated and requires multidisciplinary care. Hemophilia treatment centers (HTC) exist to provide comprehensive medical and psychosocial services to patients and their families. Soucie et al. described a survival advantage for patients with hemophilia treated at an HTC compared to those treated in alternative systems in the United States.22
Plasma-derived and recombinant factor products
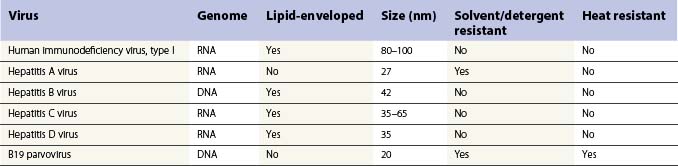
The discovery in the 1960s that factor VIII is concentrated about tenfold in cryoprecipitate, and the subsequent description of the production of antihemophilic globulin in a closed-bag system made more specific replacement therapy for people with hemophilia possible. Unfortunately, however, prior to the application of effective virucidal methods to such concentrates in the 1980s, a significant proportion of hemophilia patients contracted hepatitis C and HIV/AIDS. All factor concentrates, whether plasma derived or recombinant are now virucidally treated through viral inactivation, attenuation or elimination which has eradicated lipid-enveloped viruses such as HIV, hepatitis B and C and West Nile virus. There have been no documented cases of transmission of these diseases since 1985 for FVIII concentrates and 1990 for FIX concentrates. However, routine virucidal treatment does not reliably eradicate some nonlipid-enveloped viruses such as parvovirus B-19 and hepatitis A and outbreaks related to factor concentrate have been reported.23,24 The recommendation remains that all infants who receive factor replacement are vaccinated against hepatitis A and B during infancy. Recently, a concern has been raised that plasma derived concentrates, and even possibly recombinant factor concentrates stabilized with human albumin could theoretically transmit prions associated with Creutzfeldt–Jakob disease (CJD) or variant CJD.25
Table 34.3 shows the main blood-borne viruses with their genomic and physicochemical characteristics. It can be seen that the risk of HIV infection in virally inactivated concentrates is miniscule.26 The risk of transmission of hepatitis B and C has also been essentially erradicated. However, as mentioned, there remains the problem of possible transmission of hepatitis A and parvovirus in solvent detergent treated plasma-derived clotting factors; for this reason, many inactivation processes include more than one virucidal method. For FIX concentrate the process of nanofiltration has been used to prevent transmission of hepatitis A and parvovirus.
Although the FIX gene was cloned in 1982 the development of recombinant FIX was considerably more difficult because of the post-translational modifications required for full activity.27
A recent article provides a review of contemporary coagulation factor products and their uses in inherited disorders or coagulopathy.28
1-Deamino-8-D-arginine vasopressin (DDAVP) or desmopressin
DDAVP or desmopressin is a synthetic analog of vasopressin which lacks the vasopressor effects. It has played an important role in the treatment of mild hemophilia A and type 1 von Willebrand disease (vWD) for several decades.29 In the mid 1970s, it was demonstrated that an infusion of DDAVP increased the plasma concentrations of FVIII:C, vWF and tissue plasminogen activator (tPA) when infused into normal volunteers.30,31 The increase in plasma FVIII:C and vWF is generally two- to sixfold baseline levels. The increased plasma levels of vWF are secondary to release from Weibel–Palade bodies located in endothelial cells and perhaps also from platelet α-granules.31 The source of the FVIII store released upon treatment with DDAVP is not established.
Lethagen et al demonstrated the effectiveness of intranasal administration, which is an ideal choice for home administration.32 Intranasal DDAVP approximates the effect obtained with intravenously or subcutaneously administered product.33 For adults with mild hemophilia A the recommended dose of 0.3 µg/kg (IV or SQ), or 300 µg intranasally, can be repeated at intervals of 12–24 hours. However, tachyphylaxis (depletion of FVIII/vWF from repeated endothelial exocytosis into plasma) may develop, as well as flushing and/or hypotension. Mannucci et al. reported that the response to a second dose of DDAVP is approximately 30% less than that obtained with the first.34 It was also demonstrated that a full response to DDAVP is usually recovered within 3–4 days after a break in treatment. Since DDAVP also stimulates the release of tPA (a profibrinolytic enzyme), consideration should be given to concurrent administration of an antifibrinolytic agent with DDAVP for the management of bleeding in the oropharynx or gastrointestinal tract. DDAVP is an antidiuretic which may promote excessive free water retention and subsequent hyponatremia. It is usually therefore avoided in younger children and the elderly. It has also been suggested that DDAVP may rarely cause angina pectoris, stroke and coronary artery thrombosis in the elderly population where caution is advised.35
Antifibrinolytic agents
Antifibrinolytic agents are an often underutilized adjunctive therapy in the management of bleeding in hemophilia. These agents, ε-aminocaproic acid and tranexamic acid, act by inhibiting fibrinolysis mediated by plasmin thereby enhancing clot stability. Given their mechanism of action, they are particularly helpful in anatomic areas subject to increased fibrinolysis such as the oro- and nasopharynx and often also in menorrhagia.36
Inhibitors
Alloantibody formation against FVIII or FIX in response to treatment is now considered to be the most significant complication of hemophilia care. One of the earliest references recording inhibitors was that of Davidson et al. in 1949.37
The development of inhibitors is more common in patients with hemophilia A than in those with hemophilia B. Inhibitors are more common in patients with severe forms of hemophilia A or B. More recent studies have suggested that up to 20–30% of patients with severe hemophilia A and up to 3% of severe hemophilia B patients will develop a clinically significant inhibitor at some time in their life.38–40 Inhibitors are significantly less common in mild to moderate hemophilia A, at 3–15%.38,41,42
Prospective clinical trials evaluating recombinant FVIII products in previously untreated patients (PUP) provided invaluable insight into the incidence of inhibitors with these products in hemophilia A. Notably, these prospective trials performed frequent laboratory surveillance, which as discussed below may account for the higher incidence of inhibitors than was previously appreciated. In the Kogenate™ PUP study, 20% of patients developed an inhibitor after a median of 9 exposure days. Of those with severe disease, 25% developed an inhibitor, while less than 10% with mild or moderate disease did so. In patients who developed an inhibitor, approximately 50% had spontaneous resolution or persistently low titers despite continued treatment. The authors described these as ‘transient’ inhibitors. The cumulative probability of inhibitor development was 36% after 18 days of treatment.38 This incidence rate is similar to that reported in the Recombinate™ PUP study with a cumulative probability of 38% at a median 25 days exposure.43 As would be expected, lower rates of inhibitor development were reported in trials that enrolled previously treated patients (PTP).44,45
Inhibitors are typically IgG subclass 4 or 1, appear at a median of 9–12 days after exposure to factor concentrate, and do not naturally occur prior to factor exposure. Presumably the higher incidence of inhibitor development in patients with severe hemophilia A is explained by the almost complete absence of circulating endogenous FVIII. The absence of in utero exposure to FVIII is thus associated with a failure to develop tolerance to this antigen, such that patients remain predisposed to antibody formation upon exposure to exogenous factor later in life. Data available in the international electronic databases suggest that patients with large deletions (>200 bp) or stop mutations are more likely to develop inhibitors, while those with smaller deletions or missense mutations are less likely to do so.6,46 Patients with moderate or mild hemophilia A may synthesize FVIII that has an abnormal tertiary or quaternary structure. Epidemiologically, these individuals appear to be especially prone to develop inhibitors later in life, and particularly at times of intensive exposure to FVIII replacement, such as after surgery.47 It is hypothesized that at such times, the immunologic system, which is in a state of activation, is more likely to perceive the normal wild type FVIII as a ‘foreign’ antigen.
One such risk factor is subject race/ethnicity, which appears to influence inhibitor formation. The Malmo International Brother Study (MIBS) reported the incidence of inhibitors in Caucasian and black people to be 27% and 56% respectively.48 Hispanic people also appear to be at higher risk of inhibitor formation. A recent study has suggested that this disparity may be at least partially explained by the types of recombinant products used in these populations and their underlying FVIII haplotypes. On the basis of four single nucleotide polymorphisms (SNPs) within the FVIII protein, six wild-type haplotypes, designated H1 through H6, can be discerned. However, only the H1 and H2 haplotypes match the available recombinant factor products approved for clinical use. Viel et al. reported the background haplotypes for 78 black patients with hemophilia. Patients with H3 or H4 haplotypes (which were more prevalent in African-Americans) had a significantly higher incidence of inhibitor development than those with H1 or H2 (odds ratio, 3.6; 95% confidence interval, 1.1 to 12.3; P = 0.04).49 This study suggests that a mismatch between patient haplotype and replacement product haplotype may predisopose to development of an inhibitor. If confirmed, this study suggests that ‘individualized’ forms of FVIII replacement therapy could in theory mitigate the risk of inhibitor formation in the future.
There also appears to be a familial predisposition to inhibitor development. Siblings of patients with hemophilia and an inhibitor are at increased risk of inhibitor development.41 The results of the CANAL (Concerted Action on Neutralizing Antibodies in severe hemophilia A) cohort study led to the development of a risk stratification score that predicted the development of inhibitory antibodies in untreated patients with severe hemophilia A.50,51 The risk factors that were proposed in this scoring system were family history of inhibitors, presence of a high risk gene mutation and intensive treatment at first episode.
It has been suggested that switching between FVIII products and use of recombinant factor products is associated with higher incidence of inhibitor development. Although results from the CANAL cohort study do not support the latter association,52 a great deal of circumstantial evidence continues to raise this concern.53,54
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


