As with other enveloped viruses, the initial step in replication is binding of viral surface glycoproteins to cell surface receptors (Figure 1). All influenza viruses initiate infection by binding to terminal sialic acid residues on oligosaccharides of glycolipids and glycoproteins on the surface of host cells. Avian influenza virus strains have a preference for binding to the terminal sialic acid (Sia) with an α(2,3) linkage to galactose (Siaα2,3Gal), whereas human influenza virus strains prefer to bind to terminal sialic acids with an α(2,6) linkage (Siaα2,6Gal) (Couceiro et al., 1993; Connor et al., 1994; Ito and Kawaoka, 2000; Rogers and D’Souza, 1989; Rogers and Paulson, 1983; Rogers et al., 1983; Matrosovich et al., 2000; Matrosovich et al., 1997). This is reflected in part by the primary structure of the receptor-binding site of the HA: avian H2 and H3 viruses contain a glutamine at amino acid position 226 and a glycine at amino acid position 228, which confer efficient binding to Siaα2,3Gal. Human influenza viruses of the H2 and H3 subtypes possess leucine and serine at these positions, which are critical for efficient binding to Siaα2,6Gal. The receptor-binding specificity of H1 HAs is determined by the amino acids at positions 190 and 225, with glutamate and glycine for avian influenza viruses, and aspartate at both positions for human influenza viruses. The preference of avian and human influenza viruses for Siaα2,3Gal and Siaα2,6Gal, respectively, is likely a result of host adaptation since epithelial cells in aquatic bird intestines (where aquatic bird influenza viruses primarily replicate) express predominantly Siaα2,3Gal, whereas epithelial cells in the respiratory tract of mammals (the major site of human influenza virus replication) possess mostly Siaα2,6Gal. The differences in receptor-binding specificity between human and avian influenza viruses are most likely a critical determinant of host restriction (i.e. the inability of avian influenza viruses to efficiently replicate in humans, and vice versa). Thus, avian influenza virus HAs may have to mutate to acquire human-type receptor-binding specificity before they can cause a pandemic in humans. In fact, this was observed with the HA proteins of the pandemic 1918, 1957 and 1968 viruses, which were of avian influenza virus origin (based on their genetic make-up), but bound efficiently to Siaα2,6Gal, that is, the human-type receptor (Matrosovich et al., 2000).
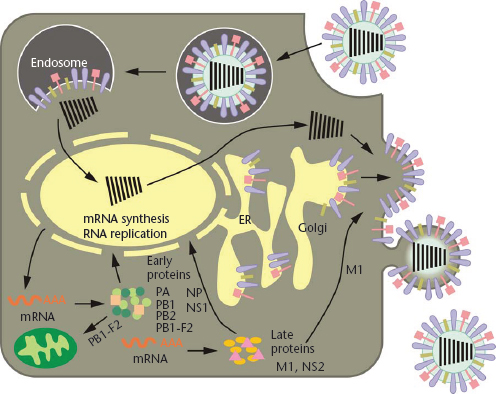
Figure 1 Schematic diagram of the influenza viral life cycle. Following receptor-mediated endocytosis, the viral ribonucleoprotein (vRNP) complexes are released into the cytoplasm and subsequently transported to the nucleus, where replication and transcription take place. mRNAs are exported to the cytoplasm for translation. Early viral proteins, that is, those required for replication and transcription, are transported back to the nucleus. Late in the infection cycle, the M1 and NS2(=NEP) proteins facilitate the nuclear export of newly synthesised vRNPs. PB1-F2 associates with mitochondria. The assembly and budding of progeny virions occurs at the plasma membrane.
Reproduced with permission from Neumann et al., 2009, Nature 459: 931–939.
Following receptor-binding, the virus is endocytosed in a coated vesicle, ultimately ending up in an endosomal compartment, where the pH is lowered and the viral membrane fuses with the endosomal membrane (Figure 1). For this to happen, the HA protein undergoes a structural rearrangement wherein the N-terminal region of the HA2 subunit, the highly conserved fusion peptide, is translocated to the head of the HA protein and near the target membrane (Bullough et al., 1994). This crucial stage of replication requires that the HA protein be already cleaved into its two subunits, HA1 and HA2, to free the fusion peptide. All highly pathogenic avian influenza viruses possess multiple basic amino acids at the HA cleavage site. By contrast, low pathogenic influenza viruses possess a single basic amino acid at this site. These viruses can replicate only in cells at mucosal surfaces where trypsin-like serine proteases are abundant. This is because the HA cleavage site of these viruses is only cleaved by these types of proteases. By contrast, the multibasic HA cleavage site of highly pathogenic avian influenza (HPAI) viruses can be cleaved by serine proteases and by intracellular membrane-bound furin-like proteases. Put simply, this means that HPAI viruses can leave the mucosal surfaces and cause systemic infections, whereas the spread of viruses of low pathogenicity is restricted. Thus, the HA cleavage sequence is a major determinant of viral pathogenicity.
Following fusion of the viral and cellular membranes, the viral ribonucleoprotein (vRNP) complex is released into the cytoplasm. The viral RNP complex is transported to the cell nucleus, where viral replication and transcription take place (Figure 1). Influenza viruses are one of only a few RNA viruses that replicate in the nucleus. To initiate mRNA synthesis, the virus cleaves the terminal 10–13 nucleotides of capped cellular mRNAs and uses them to initiate viral transcription. The viral mRNAs are exported from the nucleus and translated in the cytoplasm by the cellular protein synthesis machinery. The viral RNAs also serve as templates for the synthesis of a full-length, positive-sense copy of the viral genes, which are in turn used as templates for viral RNA synthesis. This replication step results in the amplification of the viral RNAs.
Similar to host-specific amino acids in HA, species-specific amino acids have been identified that distinguish the polymerase proteins of avian and human influenza viruses. The most prominent example is position 627 of the PB2 polymerase subunit: most avian influenza viruses possess glutamic acid at this position; by contrast, most human influenza viruses (with the exception of the pandemic (H1N1) 2009 viruses, see below) encode lysine at this position (Subbarao et al., 1993; Hatta et al., 2001). The lysine at this position confers the ability to replicate efficiently in mammalian cells, whereas viruses possessing glutamic acid at PB2-627 are restricted in their replicative efficiency in mammalian cells. The conversion from glutamic acid to lysine at position PB2-627 may thus be critical for an avian influenza virus to cause a pandemic in humans. It was therefore interesting that the virus that caused the pandemic in 2009 encodes glutamic acid at PB2-627K. Subsequent studies showed that the lack of a basic residue at position 627 can be compensated for by a basic amino acid at position 591 of the PB2 protein (Mehle and Doudna, 2009; Yamada et al., 2010). Hence, there are key amino acids in viral proteins that critically affect replication in mammalian species. However, the lack of such amino acids can be compensated for (at least in the case of PB2-627) by an alternative amino acid change.
Late in the infection cycle, newly synthesised vRNP complexes are exported to the cytoplasm with the help of the viral NS2 (=NEP, nuclear export protein) and M1 proteins. The assembly of virions, which takes place at the plasma membrane (Figure 1), is still poorly understood. Multiple interactions among viral proteins, and between viral and host proteins, are likely necessary to orchestrate virion formation. In addition, ‘packaging signals’ in the viral RNA segments are critical for the efficient incorporation of the viral gene segments into virions, although the exact role of these signals has yet to be clearly defined. Newly formed virions bud from the plasma membrane (Figure 1); their efficient release from host cells requires the sialic acid-cleavage activity of the viral NA protein.
Epizoology
The most interesting and unique aspect of the biology of influenza A viruses is the epizoological relationship with their hosts. The prevailing view is that avian influenza viruses have been fixed in the avian population for millennia, where they have adapted to selected orders, including waterfowl (Charadriiformes) and shorebirds (Anseriformes), which now serve as ‘primordial’ reservoirs for non/low-pathogenic strains (Webster et al., 1992). These non/low-pathogenic strains are thought to be perpetuated by transmission within and between flocks primarily during nesting and fledging at sites of water. The original discovery of avian influenza viruses in waterfowl (Slemons and Easterday, 1977; Pereira et al., 1965) eventually led to extensive surveillance efforts in subsequent years. The best evidence for the prevailing view may be found in the lakes of Minnesota and in Alaska (Hinshaw et al., 1979; Ito et al., 1995), where studies have shown that migrating ducks shed enough virus to be cultured directly from lake water and that virus may overwinter in these cold climates.
Phylogenetic analyses based on comparisons of viral gene sequences have identified host-specific virus lineages: typically, avian influenza virus genes form one branch, whereas human and swine influenza virus genes cluster on another branch (Webster et al., 1992; Smith et al., 2009a, b) in addition to the equine virus lineage. Further analyses have also identified host-specific amino acids, that is, amino acids at specific positions that distinguish avian, human and (in some cases) swine viruses (examples for the HA and PB2 proteins were already discussed).
It is difficult to accurately measure clinical disease in wild waterfowl, but most influenza virus infections in avian species seem to be non or low-pathogenic with relatively low rates of evolution: in essence, most avian influenza viruses appear to be well-adapted to their natural hosts. However, evolution can be measured with the highest rates being found for the HA, NA and NS1 proteins. The evolutionary rates are higher for influenza viruses circulating in poultry or mammals (compared with viruses circulating in wild waterfowl), most likely reflecting adaptive pressure. As stated earlier, avian influenza viruses typically do not cause lethal infections in wild waterfowl. Notable exceptions have occurred with highly pathogenic avian H5N1 influenza viruses; an outbreak associated with lethality occurred among exotic birds in a zoological park in Hong Kong (Sturm-Ramirez et al., 2004). In 2005, an outbreak of highly pathogenic avian H5N1 viruses killed hundreds of wild waterfowl at Qinghai Lake, China (Chen et al., 2005, 2006; Liu et al., 2005). These so-called ‘Qinghai Lake’ viruses have now spread into Europe, parts of Africa and the Middle East, where they are enzootic in poultry populations and continue to infect humans with high case fatality rates. It is currently not understood why these viruses are highly pathogenic in wild waterfowl.
When avian influenza viruses are found outside feral bird populations, the consequences can be devastating. Direct transmission of viruses with genes closely related to those found in waterfowl has been demonstrated in humans, swine, horses, seals, mink and galliform birds (chickens and turkeys). Although these transmissions have generally been nonvirulent, there have been transmissions with lethality in all of these species. For the first time, the events of the 1997 Hong Kong human H5N1 outbreak initiated public health concerns regarding purely unadapted avian strains.
Clinical Features
Infections with avian influenza viruses range from asymptomatic to lethal. One characteristic of almost all avian influenza viruses is the ability to kill chick embryos of approximately 11 days old or younger. This is in contrast to human influenza A isolates, which usually do not kill embryos. In older embryos, the capacity for lethality becomes more variable. Many nonpathogenic isolates do not kill older (≥14 days) embryos, whereas, thus far, all highly pathogenic isolates are lethal to older embryos, usually within 48h. The terms ‘low pathogenic’ and ‘highly pathogenic’ refer to the clinical features of avian influenza infection in chickens. However, influenza isolates can generate very different clinical features when tested in different species or under different conditions. Most ‘mildly pathogenic’ or nonpathogenic strains replicate only in the respiratory tract and gut of the infected bird, where they can cause standard inflammatory responses and illness or elicit no clinical signs at all.
When a virulence shift occurs during a mild infection, or when a highly pathogenic virus is introduced into chickens or turkeys, the clinical features change appreciably. The virus can now often be found in the vascular tissue, brain, spleen and external skin. Necrosis of the comb and wattle are common features, as are leg lesions. Nervous signs are noted with some HPAI strains but seem to depend upon the rapidity with which mortality occurs following infection. For example, with isolates from the 1997 H5N1 outbreak in Hong Kong, and the recent Asian H5N1 strains, little nervous involvement is noted because the study birds generally die between 12 and 32h post-infection, depending upon the route of inoculation (Suarez et al., 1998; Webster and Hulse, 2004). Severe pulmonary congestion and vascular infiltration, with high levels of replication in cardiac myocytes, are clinical hallmarks of infections with the 1997 H5N1 strains; lesions are severe but mostly limited to pulmonary and cardiovascular tissue. In contrast, highly pathogenic viruses from the H5N2 outbreak in chickens in Mexico in 1994–1995 produced a wide variety of lesions in systemic tissues and occasional nervous signs with clear virus replication in neuronal tissue (Swayne, 1997).
There are marked host-specific variations in the pathogenesis of avian influenza virus strains. Although highly pathogenic strains are so designated based upon their pathogenesis in chickens, they may exhibit few or no clinical signs in other species of birds. Recently this has become very important in the spread of highly pathogenic H5N1 viruses in Asia and elsewhere, where several species of ducks may be infected with H5N1 strains that can replicate to high titres, be shed in high concentrations, and cause disease and death of the infected animals. Other species such as ostrich and quail can also exhibit marked differences in clinical signs even though the virus replicates well. This host-specific variation can clearly be problematic for disease control in birds (see below).
Virulence Shifts
One fascinating aspect of the molecular biology of avian influenza viruses is their capacity to undergo virulence shifts following replication in naive avian populations. The first indication of this kind of shift came in Ontario, Canada in 1966, when nonpathogenic turkey viruses of the subtype H5N9 may have given rise to a highly pathogenic outbreak in turkeys. Ten years later in Victoria, Australia, a highly pathogenic H7N7 strain arose at about the same time as a nonpathogenic isolate was cultured from nearby ducks. In 1983, in the northeastern United States, H5 strains once again caused highly pathogenic outbreaks; there was strong evidence that an H5N2 nonpathogenic precursor gave rise to a highly lethal variant in commercial poultry that was subsequently contained by quarantine and slaughter. The single most important change in the transition was the loss of a carbohydrate site in the HA protein. This change appeared to have exposed a multibasic cleavage site (see Replication section) that had already appeared earlier in this strain; the multibasic cleavage site is separated from the carbohydrate attachment site by hundreds of amino acids in the primary polypeptide sequence, but these two sites are adjacent in the three-dimensional projected structure.
The first molecular evidence for conversion of low-pathogenic avian influenza (LPAI) to HPAI viruses due to the acquisition of a multibasic HA cleavage site came from the outbreak in Mexico in 1993–1995, in which several basic amino acids were inserted into the HA cleavage site of the original LPAI virus during its circulation in poultry (Table 2). One mechanism for the virulence shift appears to be a spontaneous duplication of purines at RNA sequences encoding the cleavage site, and a theoretical mechanism explaining how this might happen has been proposed (Perdue et al., 1996, 1997). Outbreaks of HPAI in Chile in 2003 (Suarez et al., 2004) and in Canada in 2004 (Pasick et al., 2005) led to the identification of another mechanism for the acquisition of high virulence, since these isolates acquired a multibasic HA cleavage site by apparent RNA–RNA recombination with other viral genes; this mechanism had been shown previously in cell culture.
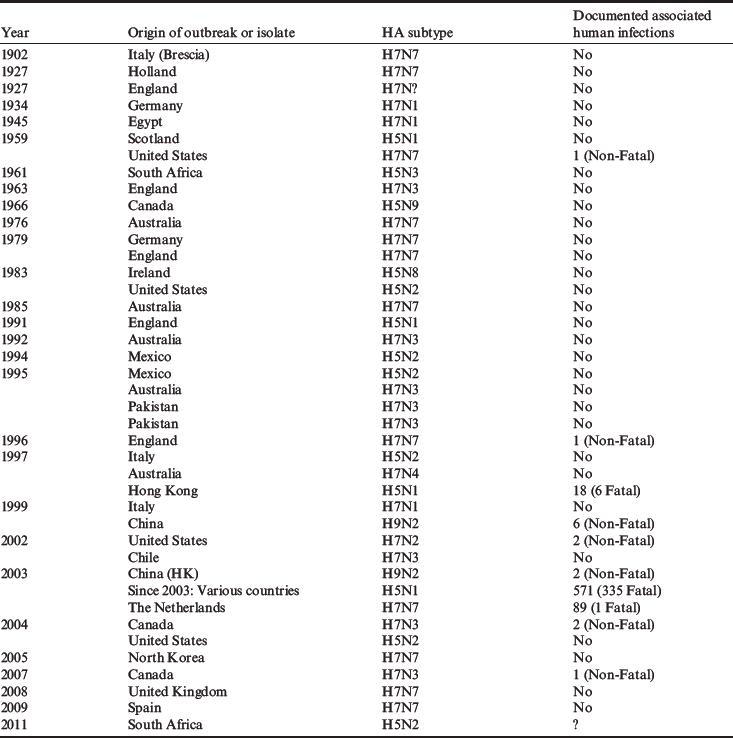
Table 2 Global history of the emergence of highly pathogenic and human infectious avian influenza viruses
Control
The best method for control of avian influenza in commercial avian operations is to employ strict biosecurity measures. The most prevalent mechanism for initiating an epizootic appears to be human-vectored mechanical transfer of virus between poultry houses or farms. Although the severe disease almost always manifests as a respiratory infection, aerosol transmission over significant distances has not been demonstrated in poultry outbreaks.
Controlling H5N1 virus outbreaks in Asia and other parts of the world (particularly the Middle East) has proven extremely difficult. Although there have been efforts to control the disease in commercial operations, the extensive backyard farming of poultry in large parts of Southeast Asia and the Middle East has presented a virtually unsolvable biosecurity problem. In addition, endemic and clinically inapparent infections in ducks and migrating waterfowl complicate the control of these viruses (see www.fao.org, www.oie.int and www.who.org for updated information on control efforts).
Vaccination works very well for generating neutralising antibodies in birds and for disease control. It has been used successfully in the American States of Minnesota and Utah against various subtypes. However, it appears to have been less effective in the H5N2 outbreak in Mexico; the exact reasons for this are unknown. Vaccination has also been effective in Italy and Chile. For review articles on experimental and approved avian influenza vaccines, see (Capua et al., 2009; Chen, 2009; Domenech et al., 2009; Fuchs et al., 2009; Koch et al., 2009); for an overview of currently available avian influenza vaccines, see ftp://ftp.fao.org/docrep/fao/011/ai326e/ai326e00.pdf.
Properly administered vaccination with concomitant biosecurity, surveillance and slaughter programmes may control avian influenza viruses. On the other hand, low vaccine coverage or vaccines that prevent recognisable disease symptoms without virus neutralisation may lead to subclinical infections and increase evolutionary pressure, resulting in antigenic escape variants. Such scenarios may have occurred during efforts to eradicate highly pathogenic avian H5N1 influenza viruses.
Antivirals are also effective, but their expense and the rapidity with which resistant mutants arise precludes their widespread use, either prophylactically or therapeutically, in birds. Control of the disease produced by avian influenza viruses would, therefore, seem to lie in clearer identification of their epizoological relationships in wildfowl. This will require an increase in animal surveillance. The application of strict and biosecure separation between wild animal sources and commercial animals will reduce the prevalence of these viruses.
Avian influenza viruses of the H5, H7 and H9 subtypes have infected humans. The case fatality rate is high for human infections with highly pathogenic avian H5N1 viruses, but appears to be very low for human infections with avian H7 or H9 viruses. Nonetheless, candidate vaccines to all three subtypes have been developed; several vaccines to highly pathogenic avian H5N1 viruses are now approved (reviewed in Prieto-Lara and Llanos-Mendez, 2010), and clinical trials have been conducted for vaccines to H7 and H9 viruses.
Avian Influenza Virus Infections in Humans
Before 1997, there were very few infections of humans with H7 subtype avian influenza viruses, including a case of laboratory-acquired keratoconjunctivitis and cases in humans that were associated with an apparent avian strain outbreak in seals. The first documented H7N7 conjunctivitis directly from birds was reported in 1995 (Banks et al., 1998) and one apparent respiratory infection in 1959 may have been due to a highly pathogenic H7N7 virus (see Perdue and Swayne, 2005). Indeed, experiments in volunteers suggested that most avian influenza viruses were unlikely to infect humans or mammals. However, the emergence of a limited number of lethal infections with the H5N1 subtype in 1997 in Hong Kong signalled an apparent new era in avian influenza ecology and epidemiology. This episode resulted in 18 documented infections with six fatalities (Claas et al., 1998; Subbarao et al., 1998). Prompt culling of chickens in Hong Kong probably terminated the human infections, but it was never clear why so many people who must have been exposed to viruses in the live-bird markets of Hong Kong were never infected. The most widespread infection of humans by avian influenza viruses in a single confined outbreak was in the spring of 2003 in the Netherlands. An H7N7 HPAI outbreak occurred on commercial poultry farms and an investigation was carried out during the outbreak to determine whether transmission of the virus from chickens to humans was occurring. More than 400 people reported health complaints with some 349 reporting conjunctivitis and 90 complaining of influenza-like illnesses. One-quarter of the conjunctivitis cases yielded the same H7N7 strains that were infecting poultry and two of the influenza-like illnesses yielded virus. Six of the influenza-like illnesses were caused by H3N2 influenza A infections meaning that these two viruses were co-circulating, although not in the same individual. One death attributed to the H7 virus was reported (Fouchier et al., 2004; Koopmans et al., 2004) and at least three contacts of documented cases yielded virus, so it is very likely that some human-to-human transmission occurred. Some serological results suggest there was a much wider spread of infection of humans during this outbreak.
As of November 29, 2011, 571 people have had documented infections with highly pathogenic H5N1 AI virus strains with 335 total deaths in 15 different countries (http://www.who.int/influenza/human_animal_interface/EN_GIP_20111129CumulativeNumberH5N1cases.pdf); over the last two years, most fatal cases have occurred in Egypt and Indonesia. Most experts believe that the number of infections of humans is probably higher, so it may be difficult to assess the actual case fatality rate. So far, however, serological information conclusively showing widespread asymptomatic or nonsevere infection with these viruses is limited. The apparently high mortality rate is the reason why so much attention has been focused on these viruses. Currently, the viral and host determinants of the high mortality and the underlying mechanisms are not fully understood.
As of the end of 2011, highly pathogenic avian H5N1 viruses continue to transmit from avian species to humans, but do not transmit efficiently among humans. There are concerns, however, that these viruses may acquire this ability through mutations or reassortment with human influenza viruses. The ability of highly pathogenic avian H5N1 influenza viruses to reassort with human influenza viruses has therefore been assessed experimentally (Jackson et al., 2009; Maines et al., 2006; Li et al., 2010). The HA and NA genes derived from an avian H5N1 virus (tested in the genetic background of a contemporary human influenza virus) did not confer the ability to transmit among ferrets (a widely accepted animal model for influenza transmission studies); conversely, a virus with the human influenza virus HA and NA genes and the remaining genes of an avian H5N1 influenza virus did not transmit among ferrets (Maines et al., 2006). It should be noted, however, that some of the human/avian reassortant viruses were more pathogenic than their parental viruses (Li et al., 2010). Overall, these experiments suggested that human/avian reassortant influenza viruses may not easily transmit among humans; rather, additional adapting mutations in one or several viral genes may be required to confer transmissibility in mammals. Avian influenza viruses may also acquire the ability to transmit among mammals through mutations in HA, with or without concomitant reassortment. A recent publication demonstrated this possibility for avian H5N1 viruses (Chen et al., 2011).
Role of Avian Influenza Viruses in Influenza Pandemics
At irregular intervals, novel influenza virus strains emerge that transmit efficiently among humans and cause outbreaks on a global scale (pandemics). These viruses transmit efficiently among human populations because they are antigenically distinct from the influenza virus strains circulating in humans in the years preceding the new pandemic and thus escape the existing antibody responses. In 1918, the devastating ‘Spanish Influenza’ was caused by an influenza H1N1 virus of avian origin (Figure 2), although the virus may have circulated in intermediate hosts (such as poultry or pigs) for several months or years before it was transmitted to humans. In 1957, reassortment between avian and human influenza viruses led to the ‘Asian Influenza’ virus which possessed H2 HA, N2 NA and PB1 genes of avian virus origin, whereas the remaining five viral gene segments were of human virus origin (Figure 2). This virus was replaced by a reassortant bearing H3 HA and PB1 genes of avian virus origin in combination with the remaining six viral genes from the formerly circulating H2N2 strains, leading to the generation of the virus responsible for the ‘Hong Kong Influenza’ in 1968 (Figure 2) (note that the PB1 genes of the 1957 and 1968 pandemic viruses were different, although they both originated from avian influenza viruses). In 1977, viruses of the H1N1 subtype reemerged that were similar to H1N1 viruses that circulated in the early 1950s (Figure 2). H1N1 and H3N2 viruses then co-circulated in human populations until 2009, when the pandemic (H1N1) 2009 virus emerged (Figure 2). This latter virus is a genetic reassortant; the PB2 and PA genes originated from North American avian viruses, the PB1 gene originated from a human H3N2 virus, the H1 HA, NP and NS genes originated from classic swine influenza viruses, and the N1 NA and M genes originated from an avian-like swine influenza virus of the Eurasian lineage (Dawood et al., 2009; Smith et al., 2009a, b; Trifonov et al., 2009). In fact, further analysis revealed that the novel pandemic virus likely resulted from a reassortment event between a Eurasian avian-like swine influenza virus (which provided the NA and M genes) and a so-called triple-reassortant swine influenza virus, which provided the remaining six segments (Figure 2). Triple-reassortant swine influenza viruses are human/avian/swine reassortants that have circulated in North American pig populations since the late 1990s. Until 2009, these triple-reassortant swine viruses had caused a small number of self-limiting infections in humans without further spread, suggesting that the NA and M genes acquired during the reassortment event contributed to the transmissibility of the pandemic variant. Influenza A viruses of the H1N1 subtype have circulated in humans since 1977; however, the HA protein of the pandemic (H1N1) 2009 virus was antigenically different from that of previously circulating human H1N1 viruses, resulting in a worldwide epidemic in 2009.
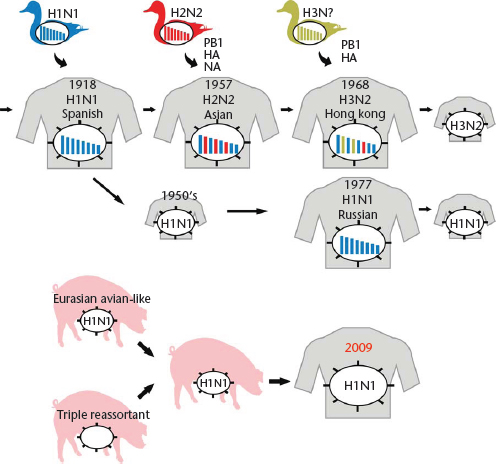
Figure 2 Emergence of pandemic influenza viruses. The ‘Spanish influenza’ was most likely caused by the transmission of an avian influenza virus to humans. In 1957, reassortment between an avian H2 and a human H1N1 virus resulted in a virus possessing avian virus H2 HA, N2 NA, and PB1 genes and the remaining genes from the human virus. Similarly, in 1968, reassortment between an avian H3 virus and a human H2N2 virus resulted in a virus possessing avian virus H3 HA and PB1 genes and the remaining genes from the human virus. In 1977, H1N1 viruses reappeared which closely resembled strains that had been circulating in the mid 1950s. The 2009 H1N1 pandemic was caused by reassortment between a triple reassortant swine virus and an Eurasian avian-like swine virus that contributed the NA and M segments to the novel pandemic virus.
Modified with permission from Neumann et al., 2009, Nature 459: 931–939.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


