Chapter 18 Infectious Diseases
Introduction to Antibiotics





Suggestions
 Know which category the individual drugs belong to, and learn the category as a whole. For example, ceftriaxone is a third-generation cephalosporin. Therefore learn about cephalosporins in general, and then know the general differences among first-, second-, third-, and fourth-generation cephalosporins. In some circumstances there might be one or two extra important bits of information pertaining to individual drugs that are important to learn.
Know which category the individual drugs belong to, and learn the category as a whole. For example, ceftriaxone is a third-generation cephalosporin. Therefore learn about cephalosporins in general, and then know the general differences among first-, second-, third-, and fourth-generation cephalosporins. In some circumstances there might be one or two extra important bits of information pertaining to individual drugs that are important to learn.



 You will have to learn “bugs” (not covered in this text) and drugs in at least three different frameworks:
You will have to learn “bugs” (not covered in this text) and drugs in at least three different frameworks:

Important Considerations
 It is required that you learn the mechanism of action (MOA) of a drug and also the mechanism of resistance, which enables an organism to live in the presence of the drug. This is important because when you are selecting an antibiotic, if your first choice does not work, you will have a better understanding of why it did not work and will be better educated to select a different antibiotic that will have a greater probability of killing the organism. For example, if a penicillin (a β-lactam) was given and the bug is known to produce β-lactamase, a third-generation cephalosporin (another β-lactam) might be a poor next choice.
It is required that you learn the mechanism of action (MOA) of a drug and also the mechanism of resistance, which enables an organism to live in the presence of the drug. This is important because when you are selecting an antibiotic, if your first choice does not work, you will have a better understanding of why it did not work and will be better educated to select a different antibiotic that will have a greater probability of killing the organism. For example, if a penicillin (a β-lactam) was given and the bug is known to produce β-lactamase, a third-generation cephalosporin (another β-lactam) might be a poor next choice. Make sure that the drug you select can get to the tissue in which the infection is located. Special examples include the following:
Make sure that the drug you select can get to the tissue in which the infection is located. Special examples include the following: Gut (intraintestinal, not intraperitoneal) infections are often best treated with oral medications that cannot be absorbed into the body (because they remain in the gut, which is where the infection is)—for example, oral vancomycin for C. difficile colitis.
Gut (intraintestinal, not intraperitoneal) infections are often best treated with oral medications that cannot be absorbed into the body (because they remain in the gut, which is where the infection is)—for example, oral vancomycin for C. difficile colitis.




Viruses are technically not alive, so antivirals are usually not referred to as antimicrobials.
Advanced Killing Techniques
Knowledge of pharmacokinetics is required to enable a clinician to be really good at knowing how to kill off an infection. Understanding some very important fundamental concepts are required (Figure 18-1).
 Minimum inhibitory concentration (MIC): The MIC of an antimicrobial is the minimum concentration that will inhibit growth of a pathogen. Obviously, it is desirable to have the concentration in the patient’s infected tissues to be greater than the MIC. If drug A has a lower MIC than drug B, then drug A kills the pathogen at a lower concentration of drug and would therefore be better at killing that particular pathogen, assuming all other factors are identical. When the MIC level of a drug for a given pathogen is low, the pathogen is considered sensitive to the drug (i.e., the drug will kill it). When the MIC is a moderate value, the pathogen’s sensitivity to the drug will be intermediate, and when the MIC is high, the pathogen will be resistant to that drug. Laboratory values often report sensitivities as S, I, or R to report sensitive, intermediate, and resistant, respectively.
Minimum inhibitory concentration (MIC): The MIC of an antimicrobial is the minimum concentration that will inhibit growth of a pathogen. Obviously, it is desirable to have the concentration in the patient’s infected tissues to be greater than the MIC. If drug A has a lower MIC than drug B, then drug A kills the pathogen at a lower concentration of drug and would therefore be better at killing that particular pathogen, assuming all other factors are identical. When the MIC level of a drug for a given pathogen is low, the pathogen is considered sensitive to the drug (i.e., the drug will kill it). When the MIC is a moderate value, the pathogen’s sensitivity to the drug will be intermediate, and when the MIC is high, the pathogen will be resistant to that drug. Laboratory values often report sensitivities as S, I, or R to report sensitive, intermediate, and resistant, respectively.
Now, exactly how the concentration of the antimicrobial stays above the MIC in the body is very important and is different for different drugs. The most important concepts are illustrated in Figure 18-1 and include:
 Time dependence is the total length of time that a drug level stays above the MIC. What matters is the total duration that the concentration is above the MIC. These drugs are called time-dependent antimicrobials. β-Lactams (penicillins, cephalosporins, and carbapenems) all fall into this category. Note that it does not matter if the drug level is just barely above the MIC or is 10 times the MIC.
Time dependence is the total length of time that a drug level stays above the MIC. What matters is the total duration that the concentration is above the MIC. These drugs are called time-dependent antimicrobials. β-Lactams (penicillins, cephalosporins, and carbapenems) all fall into this category. Note that it does not matter if the drug level is just barely above the MIC or is 10 times the MIC.
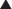
Penicillins




Moa (Mechanism of Action)





Cell Wall Destruction
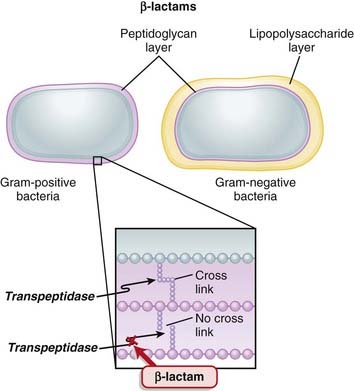
 Transpeptidases are enzymes that cross-link peptidoglycan molecules in bacterial cell walls. Cross-linking these molecules gives strength to the cell wall.
Transpeptidases are enzymes that cross-link peptidoglycan molecules in bacterial cell walls. Cross-linking these molecules gives strength to the cell wall. β-Lactams work by inhibiting transpeptidase, preventing it from forming cross-links. This results in a bacterium with a structurally deficient cell wall, typically leading to bacterial lysis (Figure 18-2).
β-Lactams work by inhibiting transpeptidase, preventing it from forming cross-links. This results in a bacterium with a structurally deficient cell wall, typically leading to bacterial lysis (Figure 18-2).





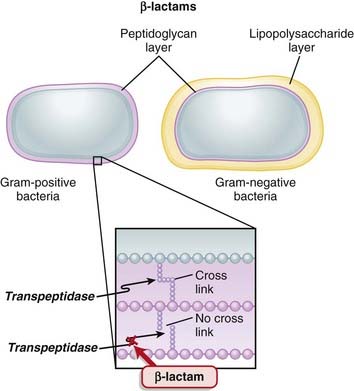
Mechanisms of Resistance
 Gram-negative bacteria, as described previously, inherently have an outer protective lipopolysaccharide layer that guards the peptidoglycan layer from attack by some β-lactams.
Gram-negative bacteria, as described previously, inherently have an outer protective lipopolysaccharide layer that guards the peptidoglycan layer from attack by some β-lactams. The main mechanisms of resistance to β-lactams include the following:
The main mechanisms of resistance to β-lactams include the following:
 Production of β-lactamase, which enzymatically destroys the four-carbon β-lactam ring
Production of β-lactamase, which enzymatically destroys the four-carbon β-lactam ring• Genes for β-lactamase may exist on plasmids or on bacterial chromosomes and may be produced constitutively (all the time) or can be induced.
• Genes that are on plasmids can readily be passed from one bacterium to another; thus resistance can be transmitted to different species.
Pharmacokinetics
 Eighty percent of penicillin is cleared by the kidneys within 4 hours. Therefore it is very quickly eliminated from the body, which is an undesirable characteristic if the goal is to expose the bacterial infection to prolonged concentrations of the drug.
Eighty percent of penicillin is cleared by the kidneys within 4 hours. Therefore it is very quickly eliminated from the body, which is an undesirable characteristic if the goal is to expose the bacterial infection to prolonged concentrations of the drug.











Important Notes
 Methicillin is no longer used clinically, because of toxicity. It is, however, used to determine whether a staphylococcal infection is sensitive to the penicillins in its class. The term methicillin-resistant Staph. aureus (MRSA) is now commonly used to describe these organisms.
Methicillin is no longer used clinically, because of toxicity. It is, however, used to determine whether a staphylococcal infection is sensitive to the penicillins in its class. The term methicillin-resistant Staph. aureus (MRSA) is now commonly used to describe these organisms.
FYI
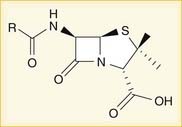
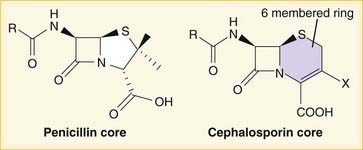
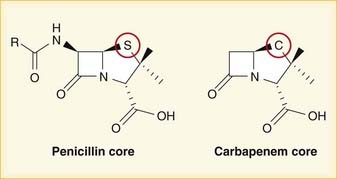
 A lactam is a chemical ring. A β-lactam is a four-molecule ring (three carbons and one nitrogen) and is the nucleus of the penicillin molecule. The penicillin nucleus is shown in Figure 18-3. Modifications to the five-membered ring are the basis on which cephalosporins and carbapenems (two other β-lactam antibiotics) were derived.
A lactam is a chemical ring. A β-lactam is a four-molecule ring (three carbons and one nitrogen) and is the nucleus of the penicillin molecule. The penicillin nucleus is shown in Figure 18-3. Modifications to the five-membered ring are the basis on which cephalosporins and carbapenems (two other β-lactam antibiotics) were derived.








Cephalosporins
Prototypes and common drugs




Moa (Mechanism of Action)





Cell Wall Destruction
 Transpeptidases are enzymes that cross-link peptidoglycan molecules in bacterial cell walls. Cross-linking these molecules gives strength to the cell wall (see Figure 18-2).
Transpeptidases are enzymes that cross-link peptidoglycan molecules in bacterial cell walls. Cross-linking these molecules gives strength to the cell wall (see Figure 18-2).


Mechanisms of Resistance
 The main mechanisms of resistance to β-lactams include the following:
The main mechanisms of resistance to β-lactams include the following:
 Production of β-lactamase, which enzymatically destroys the four-carbon β-lactam ring
Production of β-lactamase, which enzymatically destroys the four-carbon β-lactam ring• Genes for β-lactamase may exist on plasmids or on bacterial chromosomes and may be produced constitutively (all the time) or can be induced.
• Genes that are on plasmids can readily be passed from one bacterium to another; thus resistance can be transmitted to different species.










Side Effects
 Hematologic: rare cases of bone marrow suppression resulting in a low white blood cell (WBC) count (aka neutropenia or granulocytopenia)
Hematologic: rare cases of bone marrow suppression resulting in a low white blood cell (WBC) count (aka neutropenia or granulocytopenia)

Important Notes




FYI
 Note how the four-membered ring (the β-lactam) is the same as in penicillins; however, the other ring is six-membered, whereas in penicillins it is five-membered. The diagram shows the core nucleus of cephalosporins. There are currently four generations of cephalosporins (Figure 18-4).
Note how the four-membered ring (the β-lactam) is the same as in penicillins; however, the other ring is six-membered, whereas in penicillins it is five-membered. The diagram shows the core nucleus of cephalosporins. There are currently four generations of cephalosporins (Figure 18-4).



Carbapenems


Moa (Mechanism of Action)





Cell Wall Destruction
 Transpeptidases are enzymes that cross-link peptidoglycan molecules in bacterial cell walls. Cross-linking these molecules gives strength to the cell wall (Figure 18-5).
Transpeptidases are enzymes that cross-link peptidoglycan molecules in bacterial cell walls. Cross-linking these molecules gives strength to the cell wall (Figure 18-5).
Mechanisms of Resistance
 The main mechanisms of resistance to β-lactams include the following:
The main mechanisms of resistance to β-lactams include the following:
 Production of β-lactamase, which enzymatically destroys the four-carbon β-lactam ring
Production of β-lactamase, which enzymatically destroys the four-carbon β-lactam ring• Genes for β-lactamase may exist on plasmids or on bacterial chromosomes and may be produced constitutively (all the time) or can be induced.
• Genes that are on plasmids can readily be passed from one bacterium to another; thus resistance can be transmitted to different species.
• There are a few different classification schemes for β–lactamases, and there are many different individual β-lactamase enzymes. Some other names of β–lactams include penicillinase and carbapenemase.















Important Notes
 MRSA is an important resistant strain of S. aureus. It is not sensitive to carbapenems. It is treated with vancomycin.
MRSA is an important resistant strain of S. aureus. It is not sensitive to carbapenems. It is treated with vancomycin. VRE is another important resistant strain. About 50% of VRE infections are sensitive to carbapenems.
VRE is another important resistant strain. About 50% of VRE infections are sensitive to carbapenems.



FYI
 Carbapenems differ structurally from penicillins in that the five-membered ring that is attached to the β-lactam ring contains a carbon atom instead of a sulfur atom. The name carbapenem comes from carbon and penicillin. Figure 18-6 shows the core structure of a carbapenem but does not show the side chains.
Carbapenems differ structurally from penicillins in that the five-membered ring that is attached to the β-lactam ring contains a carbon atom instead of a sulfur atom. The name carbapenem comes from carbon and penicillin. Figure 18-6 shows the core structure of a carbapenem but does not show the side chains.
Glycopeptides


Moa (Mechanism of Action)
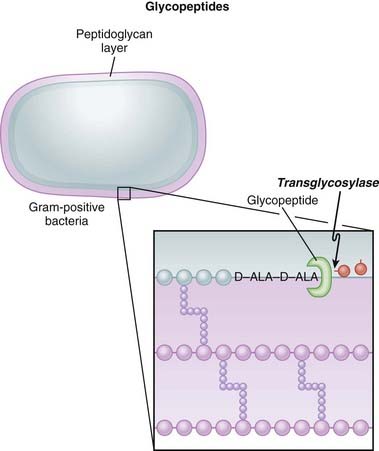
 Glycopeptides inhibit cell wall synthesis by attaching to the end of the peptidoglycan precursor units (a short four– or five–amino acid sequence called the d-alanyl-d-alanine [D-ALA-D-ALA] terminus) that are required to be laid down into the matrix. This step is catalyzed by transglycosylase. Because the glycopeptide binds to the end of the precursor, the precursor is not released from the carrier and thus peptidoglycan synthesis stops (Figure 18-7).
Glycopeptides inhibit cell wall synthesis by attaching to the end of the peptidoglycan precursor units (a short four– or five–amino acid sequence called the d-alanyl-d-alanine [D-ALA-D-ALA] terminus) that are required to be laid down into the matrix. This step is catalyzed by transglycosylase. Because the glycopeptide binds to the end of the precursor, the precursor is not released from the carrier and thus peptidoglycan synthesis stops (Figure 18-7).





Pharmacokinetics
 The half-life of vancomycin is 6 hours, whereas the half-life of teicoplanin is much longer, as high as 100 hours (assuming normal kidney function).
The half-life of vancomycin is 6 hours, whereas the half-life of teicoplanin is much longer, as high as 100 hours (assuming normal kidney function).









Side Effects
 Flushing: When vancomycin is administered quickly, the blood pressure can fall because of histamine release. Therefore vancomycin must be administered slowly (usually over 1 hour). The flushing caused by histamine release has been called red man syndrome and is not an allergy but a predictable response to rapidly administered vancomycin.
Flushing: When vancomycin is administered quickly, the blood pressure can fall because of histamine release. Therefore vancomycin must be administered slowly (usually over 1 hour). The flushing caused by histamine release has been called red man syndrome and is not an allergy but a predictable response to rapidly administered vancomycin.


Important Notes
 Although vancomycin can kill S. aureus organisms that are resistant to cloxacillin (or methicillin [i.e., MRSA]), cloxacillin has a lower MIC than vancomycin for strains that are β-lactam susceptible. Therefore cloxacillin is a better killer of S. aureus when there is no resistance. Vancomycin is not a stronger antibiotic. It is simply a different but paradoxically slightly weaker one that avoids β-lactam resistance.
Although vancomycin can kill S. aureus organisms that are resistant to cloxacillin (or methicillin [i.e., MRSA]), cloxacillin has a lower MIC than vancomycin for strains that are β-lactam susceptible. Therefore cloxacillin is a better killer of S. aureus when there is no resistance. Vancomycin is not a stronger antibiotic. It is simply a different but paradoxically slightly weaker one that avoids β-lactam resistance.




Fluoroquinolones


MOA (Mechanism of Action)
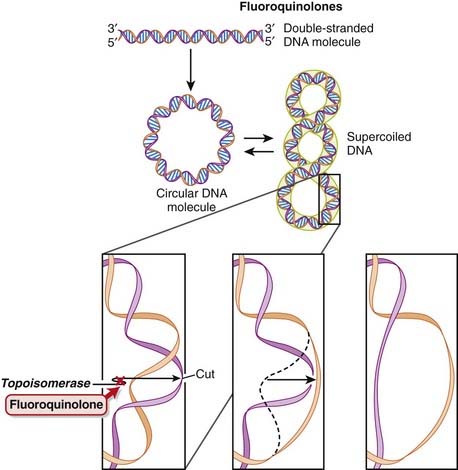
 DNA is normally supercoiled. Supercoiled DNA is under too much tension to be separated, so an extra step is required before replication and transcription can occur. DNA gyrase relaxes supercoiled DNA by cutting it, allowing rotation to occur, and then reattaching it.
DNA is normally supercoiled. Supercoiled DNA is under too much tension to be separated, so an extra step is required before replication and transcription can occur. DNA gyrase relaxes supercoiled DNA by cutting it, allowing rotation to occur, and then reattaching it. Fluoroquinolones bind to and inhibit DNA gyrase (also called topoisomerase II) and topoisomerase IV. Fluoroquinolones inhibit DNA gyrase in gram-negative organisms and topoisomerase IV in gram-positive organisms (Figure 18-8).
Fluoroquinolones bind to and inhibit DNA gyrase (also called topoisomerase II) and topoisomerase IV. Fluoroquinolones inhibit DNA gyrase in gram-negative organisms and topoisomerase IV in gram-positive organisms (Figure 18-8).




Pharmacokinetics
 Fluoroquinolones can enter human cells easily and therefore are often used to treat intracellular pathogens.
Fluoroquinolones can enter human cells easily and therefore are often used to treat intracellular pathogens.











Important Notes
 Originally, quinolones were mainly effective against gram-negative bacteria, but newer agents are useful against gram-positive cocci as well.
Originally, quinolones were mainly effective against gram-negative bacteria, but newer agents are useful against gram-positive cocci as well.


FYI
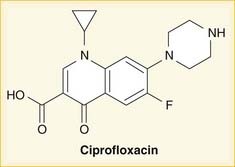
 As the name suggests, fluoroquinolones possess a fluorine ion. They all contain two six-membered rings. Ciprofloxacin is shown in the diagram (Figure 18-9). Earlier generations of fluoroquinolones were not fluorinated and were simply called quinolones. They are infrequently used now.
As the name suggests, fluoroquinolones possess a fluorine ion. They all contain two six-membered rings. Ciprofloxacin is shown in the diagram (Figure 18-9). Earlier generations of fluoroquinolones were not fluorinated and were simply called quinolones. They are infrequently used now.


Aminoglycosides


MOA (Mechanism of Action)
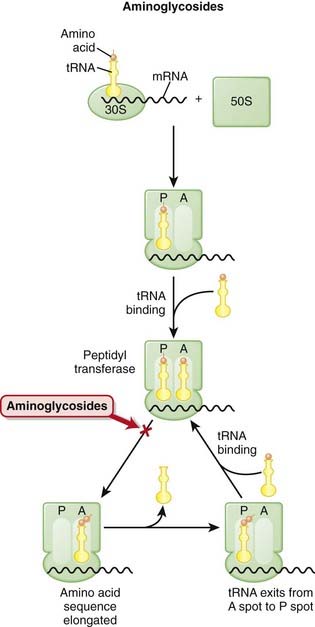
 The production of proteins from mRNA is called translation; this step requires ribosomes, transfer RNA (tRNA), and messenger RNA (mRNA) (Figure 18-10).
The production of proteins from mRNA is called translation; this step requires ribosomes, transfer RNA (tRNA), and messenger RNA (mRNA) (Figure 18-10).
 The ribosomes are different sizes:
The ribosomes are different sizes:

 There are two theories about how aminoglycosides work:
There are two theories about how aminoglycosides work:1 Aminoglycosides are protein synthesis inhibitors. They irreversibly bind the 30S ribosomal subunit (RSU). At low concentrations they cause misreading of the mRNA by ribosomes, leading to synthesis of proteins with incorrect amino acid sequences. At higher concentrations they halt protein synthesis, trapping the ribosomes at the AUG start codon. Accumulation of these abnormal initiation complexes halts translation.




Pharmacokinetics

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


