New Terminology188
The “Dose” in Dose–Response189
What Constitutes Drug Response?194
The Importance of Kinetics In Vivo199
Summary207
The ultimate therapeutic value of a molecule is determined by its effect in vivo; factors such as plasma protein binding, endogenous physiological tone and reflexes can obscure the relationship between free drug concentration and observed effect. This chapter centers on the various pharmacokinetic and pharmacodynamic factors involved in the production of in vivo drug response. The response to a drug may be observed directly or through a surrogate such as a biochemical biomarker. In addition, there may be temporal dissociation between the actual biochemical events initiated by the drug and the eventual lasting therapeutic response. Since in vivo systems are open systems, response must be linked with the time of exposure to the drug. Specifically, the kinetics of drug movement within the body are immensely important to the in vivo activity of drugs either through restricted diffusion to and from the compartments containing the therapeutic target or through the actual rate of dissociation of the drug from the target itself. Since the body is an open system (not in equilibrium), the kinetics of interaction of drugs with targets becomes paramount and a major determinant of overall in vivo activity.
Keywords
biomarkers, target coverage.
Whole Body Drug Response
The tremendous technological advances over the past 200 years in medicine and pharmacology overshadow the equally tremendous history of empirical drug discovery done in vivo over the past 5000 years. Considering ideas set forth in the Ebers papyrus (a very early source of ancient “prescriptions;” see Box 9.1), the modern period of in vitro testing comprises only 4–5% of the time empirical medicine has been practiced; the other 95–96% of the effort has been drug discovery done in vivo. Within this period the trial and error testing of herbal remedies forms a rich history of early drug discovery (e.g. see Withering and the Discovery of Digitalis; Box 9.2). In addition to observing desired therapeutic activity, the clinical testing of new drugs has led to observation of “side-effects” some of which provided insights into therapies for different diseases (see Box 9.3). With this history as a backdrop, modern pharmacology and drug discovery combines the rigor of in vitro testing (where drug concentration is known) and pharmacokinetic modeling of drug response in vivo. All the previous chapters are aimed at methods and strategies to obtain a molecule that will be therapeutically useful in vivo. This chapter considers the two topics of pharmacodynamics (primary activity at the therapeutic target) and pharmacokinetics (delivery of a therapeutic dose in vivo to the target organ) and combines them to discuss the final in vivo activity of a drug. The third main element of discovery, namely safety pharmacology, will be discussed in the next chapter.
Box 9.1
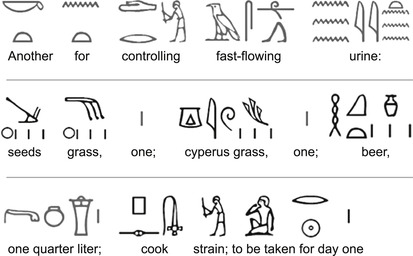
The Ebers papryus (so called because it was purchased in Luxor by Georg Ebers in 1873) is one of the oldest surviving medical records known. Thought to be written in approximately 1500 bc, it contains nearly 700 “prescriptions” (some dating from 3000 bc) for the treatment of maladies. Although many of these prescriptions are magical formulas and remedies, some hold kernels of pharmacological logic. For example, for night blindness, the “liver of ox” was suggested. Since night blindness can be caused by a deficiency in Vitamin A, and since liver is an excellent source of Vitamin A, this prescription should produce a useful effect. A partial translation of the Ebers papryus by Carpenter and colleagues[8] gives an intriguing example of ancient in vivo pharmacology.
 |
Box 9.2
While working at Birmingham General Hospital in 1779, the British physician William Withering noticed a woman with “cardiac dropsy” (fluid swelling from congestive heart failure) improve remarkably after ingesting a local herbal remedy. He learned about the remedy from an old woman herbalist and went on to determine that the active ingredient was digitalis. In the years following he systematically explored different preparations (gathered at various times of the year) to document 156 cases of the use of digitalis. In 1785 he published his famous paper “An Account of the Foxglove and Some of its Medical Uses.” This treatise describes clinical trials and notes on the therapeutic effect of digitalis and its toxicity; it is a classic chronicle of in vivo drug discovery and development. This subject became a topic of dissention when a physician colleague of Withering, Erasmus Darwin, was called for a second opinion and published the paper “An Account of the Successful Use of Foxglove in Some Dropsies and in Pulmonary Consumption.” This led them to become estranged and led to a bitter argument over what Withering considered academic plagiarism.


Box 9.3
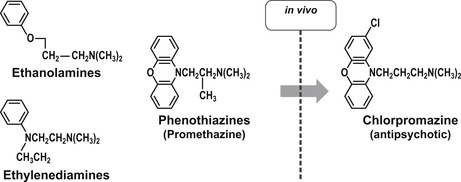
One of the main problems in drug discovery is the need for drug-like properties and adequate safety in molecules to allow testing in humans for proof of concept. When a drug meets these criteria it can be an extremely useful tool for the exploration of other possible useful activities through the observation of secondary effects (so called “side-effects”). Thus, the observed diuresis with the antibacterial sulfanilamide led to the development of the diuretic furosemide. Similarly, the development of the antidiabetic tolbutamide arose from observations with the antibacterial carbutamide. One of the most difficult areas of drug development is in diseases of the central nervous system. This is because of the subjectivity and complexity involved in the interpretation of CNS effects for diseases such as depression and psychosis. In the 1950s, efforts to develop antihistamines led to compounds that caused patients to be “disinterested in their surroundings.” This CNS side-effect was recognized as possibly of value to patients who have constant unwanted sensory input in schizophrenia. This led to the development of chlorpromazine, the first drug for that disorder.
 |
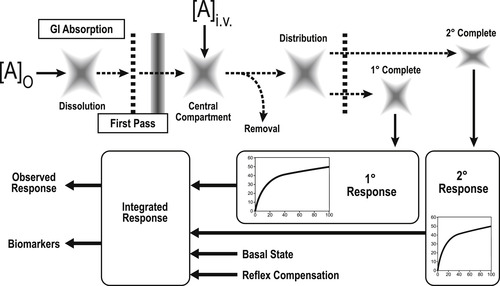
When a drug enters the body, it encounters a complex and dynamic system that has its own basal activity combined with reflex mechanisms designed to counter any perturbation. As seen in Fig. 9.1, a drug introduced orally must dissolve in the gastrointestinal tract, be absorbed through the lumen and pass through the liver before it can enter the central compartment. Drugs introduced intravenously already gain access to the central compartment. From there, drugs can be destroyed by enzymatic degradation, bound by plasma proteins, distributed to other compartments of the body, be further destroyed by hepatic metabolism or be directly excreted by the renal (or other) system. During this process, quantities of drug in body compartments can interact with tissues to produce pharmacologic responses. Some of these may be beneficial (therapeutic response) and others may counter the therapeutic effect or produce other effects that may compromise normal cellular activity. These latter interactions can raise safety issues. In general, the observed response to a drug takes into consideration the pharmacokinetic factors that can affect the magnitude of the dose given, the integration of all the responses produced by the drug, the effect of body reflexes and finally, the interaction of the drug with the ongoing basal physiological activity of the system.
New Terminology
• Biomarkers: An observed finding that can be associated with drug effect, disease or physiological state. These can be physiological readings, chemical or biochemical substances, or images.
• Target coverage: A term that encompasses the degree to which and the length of time a biological target is associated with (bound to) a drug molecule.
The “Dose” in Dose–Response
Chapter 7 and Chapter 8 extensively consider the properties required of a molecule to reach the target organ in the body and be present there for a sufficient length of time to be therapeutically useful. There are examples of where pharmacokinetics can completely dominate how a drug works in vivo (see Box 9.4). This chapter will now relate the pharmacokinetically available dose with observed in vivo response. It should be recognized that drug levels are measured in the body from samples taken from the central compartment and these may be quite different from the drug levels in the compartment actually containing the biological target for therapy (see Fig. 9.2). There are factors in the central and therapeutic compartment which can alter the concentration of drug producing an observed effect; two of these are plasma protein binding and restricted diffusion.
Box 9.4
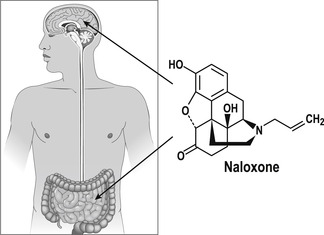
The route of administration of a drug in vivo can be a means of increasing the delivery of the drug to a given organ to optimize the active concentration at the therapeutic target. It can also effectively reduce the side-effects to a drug in vivo. For example, β-adrenoceptor bronchodilators in asthma reach the constricted bronchioles at a maximal concentration through aerosol and then dissipate through the body to arrive at the heart (a major organ for side-effects) at a much lower concentration. The opioid receptor antagonist naloxone can actually yield completely different clinical profiles when given by different routes in vivo. When administered intravenously for opiate overdose, the drug gains access to the brain where it is needed. If given via the oral route it acts exclusively on bowels to treat constipation during pain therapy without affecting the central pain-reducing effects of opiates.
 |
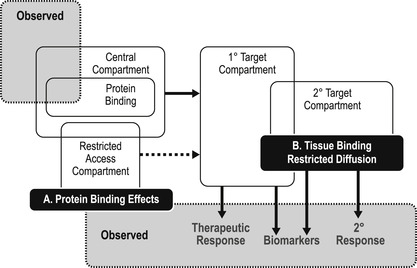
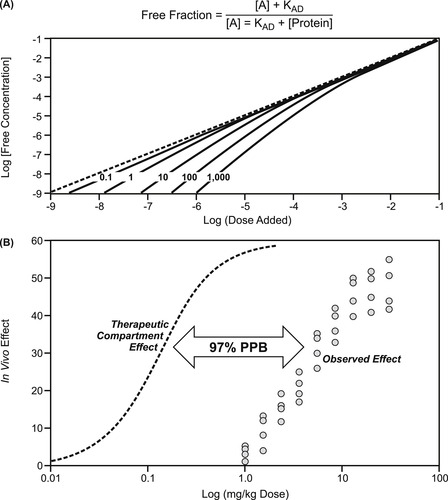
Plasma protein binding was discussed in Chapter 7 as a factor causing divergent values for dosages given in vivo and those that are actually free to produce a response. While not an extremely important factor in candidate selection of molecules, it can obscure whole body pharmacokinetics (see Box 9.5). It is also important when clinical dose is compared to observed response and the true potency (and safety margin) of drugs needs to be calculated. Plasma protein binding is a saturable process; therefore its importance may change with dosage level. Figure 9.3A shows the aberration of central compartment drug levels by plasma protein binding. It can be seen that as the drug level saturates the amount of protein available, the central compartment concentration approaches the dosage given. Depending on the amount of protein and concentration of drug, the dosage level at which this occurs varies. As seen in Fig. 9.3B, the observed response to a drug that is 97% protein bound occurs at concentrations considerably greater than the true free drug concentration, since only 3% of the added drug is free to produce a physiological response.
Box 9.5
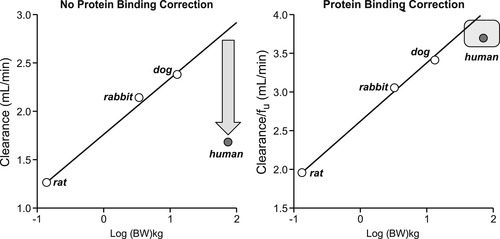
While plasma protein binding (PPB) may not seriously alter drug development strategy (see Box 8.1), it should not be ignored when pharmacokinetic profiles of compounds and/or quantitative relationships between dose and response in vivo are required. The figure below shows the difference in the veracity of predictions for human clearance made through allometric scaling. When PPB is not measured and corrected for, the regression for the α1A-adrenoceptor antagonist tamsulocin shows how the observed value falls considerably short of the allometric projection. This difference is greatly reduced when corrections are made for free drug concentration (data drawn from [9]).
 |
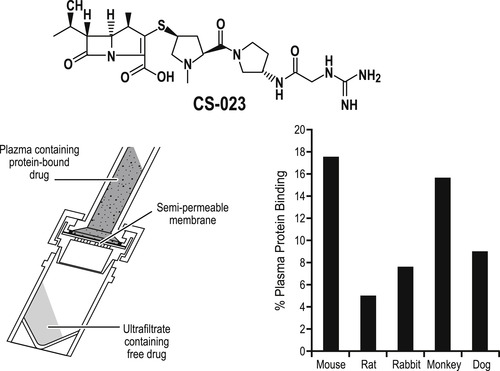
Plasma protein binding can be species-dependent; therefore it is important to quantify from actual in vivo samples to normalize dosing data. Figure 9.4 shows a convenient apparatus for this; plasma samples are placed into a centrifuge tube separated into compartments by a semi-permeable membrane. This allows aqueous media and dissolved free drug to pass to the bottom layer and protein (and protein-bound drug) to be retained on the membrane after centrifugation. Thus, plasma samples from in vivo experiments can be rapidly assessed for free drug for evaluation of true dose–response sensitivity. Shown in this figure is the 350% variation in plasma protein binding of the antibiotic CS-023 with species. [1]
 |
| Figure 9.4 (data from [1]) |
If the therapeutic compartment (containing the target of interest for primary activity) has restricted diffusion for entry, then differences between drug levels in the central compartment and the therapeutic compartment (beyond those caused by plasma protein binding) can be further exacerbated. In such a compartment the rate at which the drug achieves steady-state concentration at the biological target is given by:
(9.1)
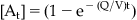
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


