The RET gene is expressed in many tissues of the body and is required for normal embryonic development of autonomic ganglia and kidney. It is unclear why germline activating mutations in this proto-oncogene result in a particular cancer of distinct histological types restricted to specific tissues, whereas other tissues in which the oncogene is expressed do not develop tumors. Interestingly, RET is the same gene implicated in Hirschsprung disease (Case 22) (see Chapter 8), although those mutations are usually loss-of-function, not activating, mutations. There are, however, some families in which the same mutation in RET can act as an activated oncogene in some tissues (such as thyroid) and cause MEN2A, while not having sufficient function in other tissues, such as the developing enteric neurons of the gastrointestinal tract, resulting in Hirschsprung disease. Thus even the identical mutation can have different effects on different tissues.
The Two-Hit Theory of Tumor Suppressor Gene Inactivation in Cancer
As introduced earlier, whereas the proteins encoded by proto-oncogenes promote cancer when activated or overexpressed, mutations in TSGs contribute to malignancy by a different mechanism, the loss of function of both alleles of the gene. The products of many TSGs have now been isolated and characterized, some of which are presented in Table 15-2.

The existence of TSG mutations leading to cancer was proposed some five decades ago to explain why certain tumors can occur in either hereditary or sporadic forms (Fig. 15-6; see discussion later in this section). It was suggested that the hereditary form of the childhood cancer retinoblastoma (see next section) might be initiated when a cell in a person heterozygous for a germline mutation in the retinoblastoma TSG, required to prevent the development of the cancer, undergoes a second, somatic event that inactivates the other retinoblastoma gene allele. As a consequence of this second somatic event, the cell loses function of both alleles, giving rise to a tumor. In the sporadic form of retinoblastoma, both alleles are also inactivated, but in this case, the inactivation results from two somatic events occurring in the same cell.
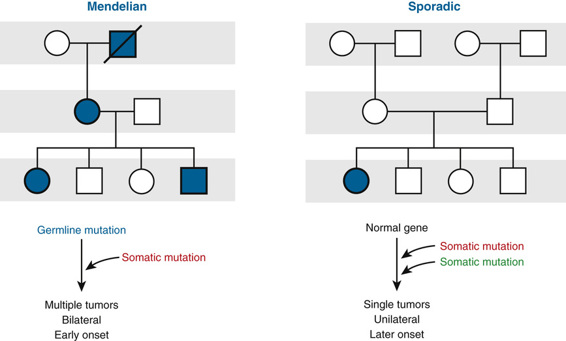
This so-called two-hit model is now widely accepted as the explanation for many hereditary cancers in addition to retinoblastoma, including familial polyposis coli, familial breast cancer, neurofibromatosis type 1 (NF1), Lynch syndrome, and Li-Fraumeni syndrome.
Tumor Suppressor Genes in Autosomal Dominant Cancer Syndromes
Retinoblastoma
Retinoblastoma is the prototype of diseases caused by mutation in a TSG and is a rare malignant tumor of the retina in infants, with an incidence of approximately 1 in 20,000 births (Fig. 15-7) (Case 39). Diagnosis of a retinoblastoma must usually be followed by removal of the affected eye, although smaller tumors, diagnosed at an early stage, can be treated by local therapy so that vision can be preserved.
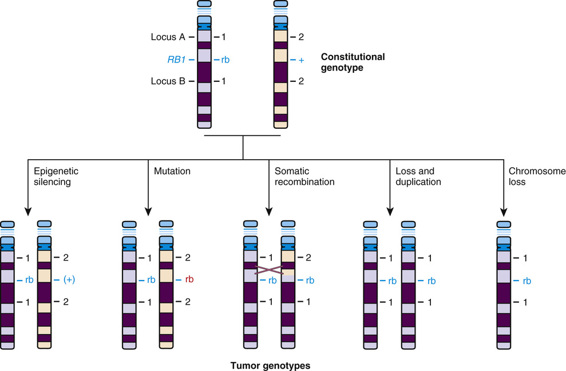
Approximately 40% of cases of retinoblastoma are of the heritable form, in which the child (as just discussed and as represented generally by the family shown in Figure 15-6) inherits one mutant allele at the retinoblastoma locus (RB1) through the germline from either a heterozygous parent or, more rarely, from a parent with germline mosaicism for an RB1 mutation (see Chapter 7). In these children, retinal cells, which like all the other cells of the body are already carrying one inherited defective RB1 allele, suffer a somatic mutation or other alteration in the remaining normal allele, leading to loss of both copies of the RB1 gene and initiating development of a tumor in each of those cells (Fig. 15-8).
The disorder appears to be inherited as a dominant trait because the large number of primordial retinoblasts and their rapid rate of proliferation make it very likely that a somatic mutation will occur as a second hit in one or more of the more than 106 retinoblasts already carrying an inherited RB1 mutation. Because the chance of a second hit is so great, it occurs frequently in more than one cell, and thus heterozygotes for the disorder often have tumors arising at multiple sites, such as multifocal tumors in one eye, in both eyes (bilateral retinoblastoma), or in both eyes, as well as in the pineal gland (referred to as “trilateral” retinoblastoma). It is worth emphasizing, however, that the occurrence of a second hit is a matter of chance and does not occur 100% of the time; the penetrance of retinoblastoma therefore, although greater than 90%, is not complete.
The other 60% of cases of retinoblastoma are sporadic; in these cases, both RB1 alleles in a single retinal cell have been mutated or inactivated independently by chance, and the child does not carry an RB1 mutation inherited through the germline. Because two hits in the same cell is a statistically rare event, there is usually only a single clonal tumor, and the retinoblastoma is found at one location (unifocal) in one eye only. Unilateral tumor is no guarantee that the child does not have the heritable form of retinoblastoma, however, because 15% of patients with the heritable type develop a tumor in only one eye. Another difference between hereditary and sporadic tumors is that the average age at onset of the sporadic form is in early childhood, later than in infants with the heritable form (see Fig. 15-6), reflecting the longer time needed on average for two mutations, rather than one, to occur.
In a small percentage of patients with retinoblastoma, the mutation responsible is a cytogenetically detectable deletion or translocation of the portion of chromosome 13 that contains the RB1 gene. Such chromosomal changes, if they also disrupt genes adjacent to RB1, may lead to dysmorphic features in addition to retinoblastoma.
Nature of the Second Hit.
Typically, for retinoblastoma as well as for the other hereditary cancer syndromes, the first hit is an inherited mutation, that is, a change in the DNA sequence. The second hit, however, can be caused by a variety of genetic, epigenetic, or genomic mechanisms (see Fig. 15-8); although it is most often a somatic mutation, loss of function without mutation, such as occurs with epigenetic silencing (see Chapter 3), has also been observed in some cancer cells. Although a number of mechanisms have been documented, the common theme is loss of function of RB1. The RB1 gene product, p110 Rb1, is a phosphoprotein that normally regulates entry of the cell into the S phase of the cell cycle (see Chapter 2). Thus loss of the RB1 gene and/or absence of the normal RB1 gene product (by whatever mechanism) deprives cells of an important checkpoint and allows uncontrolled proliferation (see Table 15-2).
Loss of Heterozygosity.
In addition to mutations and epigenetic silencing, a novel genomic mechanism was uncovered when geneticists made an unusual but highly significant discovery when they compared DNA polymorphisms at the RB1 locus in DNA from normal cells to those in the retinoblastoma tumor from the same patient. Individuals with retinoblastoma who were heterozygous at polymorphic loci flanking the RB1 locus in normal tissues (see Fig. 15-8) had tumors that contained alleles from only one of their two chromosome 13 homologues, revealing a loss of heterozygosity (LOH) in tumor DNA in and around the RB1 locus. Furthermore, in familial cases, the retained chromosome 13 markers were the ones inherited from the affected parent, that is, the chromosome with the abnormal RB1 allele. Thus, in these cases, LOH represents the second hit of the remaining allele. LOH may occur by interstitial deletion, but there are other mechanisms as well, such as mitotic recombination or monosomy 13 due to nondisjunction (see Fig. 15-8).
LOH is the most common mutational mechanism by which the function of the remaining normal RB1 allele is disrupted in heterozygotes, although each of the mechanisms shown in Figure 15-8 have been documented in different patients. LOH is a feature of tumors in a number of cancers, both heritable and sporadic, and is often considered evidence for the existence of a TSG in the region of LOH.
Familial Breast Cancer due to Mutations in BRCA1 and BRCA2
Breast cancer is common. Among all cases of this disease, a small proportion (≈3% to 5%) appears to be due to a highly penetrant dominantly inherited mendelian predisposition that increases the risk for female breast cancer fourfold to sevenfold over the 12% lifetime risk observed in the general female population. In these families, one often sees features characteristic of hereditary (as opposed to sporadic) cancer: multiple affected individuals in a family, earlier age at onset, frequent multifocal, bilateral disease or second independent primary breast tumor, and second primary cancers in other tissues such as ovary and prostate.
Although a number of genes in which mutations cause highly penetrant mendelian forms of breast cancer have been discovered from family studies, the two genes responsible for the majority of all hereditary breast cancers are BRCA1 and BRCA2 (Case 7). Together, these two TSGs account for approximately one half and one third, respectively, of autosomal dominant familial breast cancer. Numerous mutant alleles of both genes have now been catalogued. Mutations in BRCA1 and BRCA2 are also associated with a significant increase in the risk for ovarian and fallopian duct cancer in female heterozygotes. Moreover, mutations in BRCA2 and, to a lesser extent, BRCA1, also account for 10% to 20% of all male breast cancer and increase the risk for male breast cancer ten to sixtyfold over the 0.1% lifetime risk observed among males in the general population (Table 15-3).
TABLE 15-3
Lifetime Cancer Risks in Carriers of BRCA1 or BRCA2 Mutations Compared to the General Population

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


