Figure 14-1. Impulsive–compulsive disorder construct. Impulsivity and compulsivity are seen in a wide variety of psychiatric disorders. Impulsivity can be thought of as the inability to stop the initiation of actions and involves a brain circuit centered on the ventral striatum, linked to the thalamus (T), to the ventromedial prefrontal cortex (VMPFC), and to the anterior cingulate cortex (ACC). Compulsivity can be thought of as the inability to terminate ongoing actions and hypothetically is centered on a different brain circuit, namely the dorsal striatum, thalamus (T), and orbitofrontal cortex (OFC). Impulsive acts such as drug use, gambling, and obesity can eventually become compulsive due to neuroplastic changes that engage the dorsal habit system and theoretically cause impulses in the ventral loop to migrate to the dorsal loop.
| Obsessive–compulsive related spectrum disorders | Substance/behavioral addictions | Disruptive/impulse control | Sexual |
|---|---|---|---|
| Obsessive–compulsive disorder (OCD) Hair pulling (trichotillomania; TTM) Skin picking Body dysmorphic disorder (BDD) Hoarding Tourette’s syndrome/tic disorders Stereotyped movement disorders Autism spectrum disorders Hypochondriasis Somatization | Drug addiction Gambling Internet addiction Food addiction (binge eating, obesity) Compulsive shopping | Pyromania Kleptomania Intermittent explosive disorder Impulsive violence Borderline personality disorder Self harm/parasuicidal behavior Antisocial behavior Conduct disorder Oppositional defiant disorder Mania ADHD | Hypersexual Paraphilias |
Here we review the hypothetical shared neurobiology of impulsive–compulsive disorders and discuss the treatments that are available for some of these conditions. Although serotonergic treatments of OCD are well known, psychopharmacologists have generally been reluctant to embrace therapeutics for substance abuse, whereas family physicians and subspecialty substance abuse experts use the available psychopharmacological treatments more frequently. Perhaps the lack of highly effective psychopharmacologic treatments for many impulsive–compulsive disorders has led to a certain amount of therapeutic nihilism towards psychopharmacologic approaches to these conditions. Nevertheless, an explosion of neurobiological understanding of the symptom dimensions of impulsivity and compulsivity now sets the stage for novel therapeutic interventions to be discovered in the future, making it worthwhile understanding contemporary neurobiological formulations of addiction, compulsions, and impulsivity. Full clinical descriptions and formal criteria for how to diagnose the numerous known diagnostic entities should be obtained by consulting standard reference sources.
Overview of impulsive–compulsive disorders
Impulsivity and compulsivity are proposed as endophenotypes, namely symptoms linked to specific brain circuits and that are present trans-diagnostically as a dimension of psychopathology that cuts across many psychiatric disorders (Table 14-1, Figure 14-1). Simply put, impulsivity and compulsivity are both symptoms that result from the brain having a hard time saying “no.” In fact, these two symptom constructs can perhaps be best differentiated by how they both fail to control responses: impulsivity as the inability to stop initiating actions, and compulsivity as the inability to terminate ongoing actions. Both impulsivity and compulsivity are therefore forms of cognitive inflexibility.
More precisely, impulsivity is defined as acting without forethought; the lack of reflection on the consequences of one’s behavior; the inability to postpone reward with preference for immediate reward over more beneficial but delayed reward; a failure of motor inhibition, often choosing risky behavior; or (less scientifically) lacking the willpower not to give in to temptations (see definitions in Table 14-2).
| Abuse | Self-administration of any drug in a culturally disapproved manner that causes adverse consequences. |
| Addiction | A behavioral pattern of drug abuse characterized by overwhelming involvement with the use of a drug (compulsive use), the securing of its supply, and a high tendency to relapse after discontinuation. |
| Compulsivity | Repetitive actions inappropriate to the situation that persist, that have no obvious relationship to the overall goal, and that often result in undesirable consequences; behavior that results in perseveration in responding in the face of adverse consequences; perseveration in responding in the face of incorrect responses in choice situations or persistent reinitiation of habitual acts. |
| Cross-tolerance and cross-dependence | The ability of one drug to suppress the manifestations of physical dependence produced by another drug and to maintain the physically dependent state. |
| Dependence | The physiological state of adaptation produced by repeated administration of certain drugs such as alcohol, heroin, and benzodiazepines when they are abruptly discontinued, and are associated with physical drug withdrawal distinct from the motivational changes of acute withdrawal and protracted abstinence, which is part of addiction. |
| Habit | Responses triggered by environmental stimuli regardless of the current desirability of the consequences. This conditioned response to a stimulus has been reinforced and strengthened either by past experience with reward (positive reinforcement) or by the omission of an aversive event (negative reinforcement). |
| Impulsivity | The tendency to act prematurely without foresight; actions which are poorly conceived, prematurely expressed, unduly risky, or inappropriate to the situation and that often result in undesirable consequences; predisposition toward rapid, unplanned responses to internal and external stimuli without regard for the negative consequences of those reactions to themselves or others. Impulsivity is often measured in two domains: the choice of a small, immediate reward over a larger delayed reward, or the inability to inhibit behavior to change the course of action or to stop a response once it is initiated. |
| Rebound | The exaggerated expression of the original condition sometimes experienced by patients immediately after cessation of an effective treatment. |
| Reinforcement | The tendency of a pleasure-producing drug to lead to repeated self-administration. |
| Relapse | The reoccurrence, upon discontinuation of an effective medical treatment, of the original condition from which the patient suffered. |
| Tolerance | Tolerance has developed when, after repeated administration, a given dose of a drug produces a decreased effect, or, conversely, when increasingly larger doses must be administered to obtain the effects observed with the original use. |
| Withdrawal | The psychologic and physiologic reactions to abrupt cessation of a dependence-producing drug. |
On the other hand, compulsivity is defined as actions inappropriate to the situation but which nevertheless persist, and which often result in undesirable consequences. In fact, compulsions are characterized by the inability to adapt behavior after negative feedback. Habits are a type of compulsion, and can be seen as responses triggered by environmental stimuli regardless of the current desirability of the consequences of that response (Table 14-2). Habits can be seen as conditioned responses (such as drug seeking, food seeking, gambling) to a conditioning stimulus (such as being around people or places or items associated with drugs, food, or gambling in the past) that have been reinforced and strengthened either by past experience with reward (positive reinforcement) or with the omission of an aversive event (loss of the negative reinforcement that comes from withdrawal or craving). Whereas goal-directed behavior is mediated by knowledge of and desire for the consequences, in contrast, habits are controlled by external stimuli through stimulus–response associations that are stamped into brain circuits through behavioral repetition and formed after considerable training, can be automatically triggered by stimuli, and are defined by their insensitivity to their outcomes. Given that goal-directed actions are relatively cognitively demanding, for daily routines it can be adaptive to rely on habits that can be performed with minimal conscious awareness. However, habits can also represent severely maladaptive perseveration of behaviors (Figure 14-1, Table 14-1).
Neurocircuitry and the impulsive–compulsive disorders
Why can’t impulses and compulsions be stopped in various psychiatric disorders (Table 14-1, Figure 14-1)? The answer may lie in a problem in cortical circuits that normally suppress these behaviors. An oversimplification of this notion is that impulsivity and compulsivity are hypothetically neurobiological drives that are “bottom-up,” with impulsivity coming from the ventral striatum, compulsivity coming from the dorsal striatum, and different areas of prefrontal cortex acting “top-down” to suppress these drives (Figures 14-1 through 14-3). Inhibitory volitional control is thus exerted top-down by cortical mechanisms, implying that impulsivity and compulsivity could result from a relaxation of this control. According to this formulation of impulsivity and compulsivity, behavioral output is thus controlled by a balance between dual and sometimes competing neurobehavioral systems. What actually happens depends upon the balance between “top-down” and “bottom-up,” with both impulsivity and compulsivity being either caused by a failure of response inhibition systems (i.e., inadequate top-down cognitive control: Figures 14-1 through 14-3) or the result of too much pressure coming bottom-up from the ventral striatum for impulsivity (Figure 14-2) or from the dorsal striatum for compulsivity (Figure 14-3).
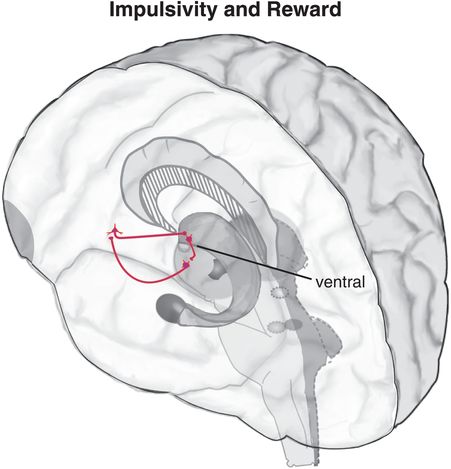
Figure 14-2. Circuitry of impulsivity and reward. The “bottom-up” circuit that drives impulsivity (shown in pink) is a loop with projections from the ventral striatum to the thalamus, from the thalamus to the ventromedial prefrontal cortex (VMPFC), and from the VMPFC back to the ventral striatum. This circuit is usually modulated “top-down” from the prefrontal cortex (PFC). If this top-down response inhibition system is inadequate or is overcome by activity from the bottom-up ventral striatum, impulsive behaviors may result.
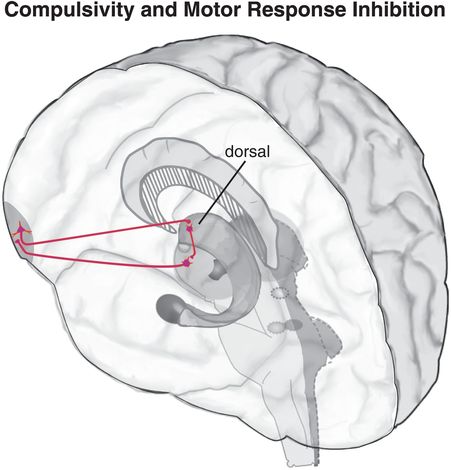
Figure 14-3. Circuitry of compulsivity and motor response inhibition. The “bottom-up” circuit that drives compulsivity (shown in pink) is a loop with projections from the dorsal striatum to the thalamus, from the thalamus to the orbitofrontal cortex (OFC), and from the OFC back to the dorsal striatum. This habit circuit can be modulated “top-down” from the OFC, but if this top-down response inhibition system is inadequate or is overcome by activity from the bottom-up dorsal striatum, compulsive behaviors may result.
Neuroanatomically, impulsivity and compulsivity are seen as engaging different neuronal loops: impulsivity as an action–outcome ventrally dependent learning system (Figure 14-2) and compulsivity a habit system that is dorsal (Figure 14-3). Many behaviors start out as impulses in the ventral loop of reward and motivation (Figure 14-2). Over time, however, some of these behaviors migrate dorsally (Figure 14-3) due to a cascade of neuroadaptations and neuroplasticity that engage the habit system by means of which an impulsive act eventually becomes compulsive (Figure 14-1). These spirals of information from one neuronal loop to another also appear to involve regulatory input from hippocampus and amygdala and other areas of prefrontal cortex (Figure 14-4).
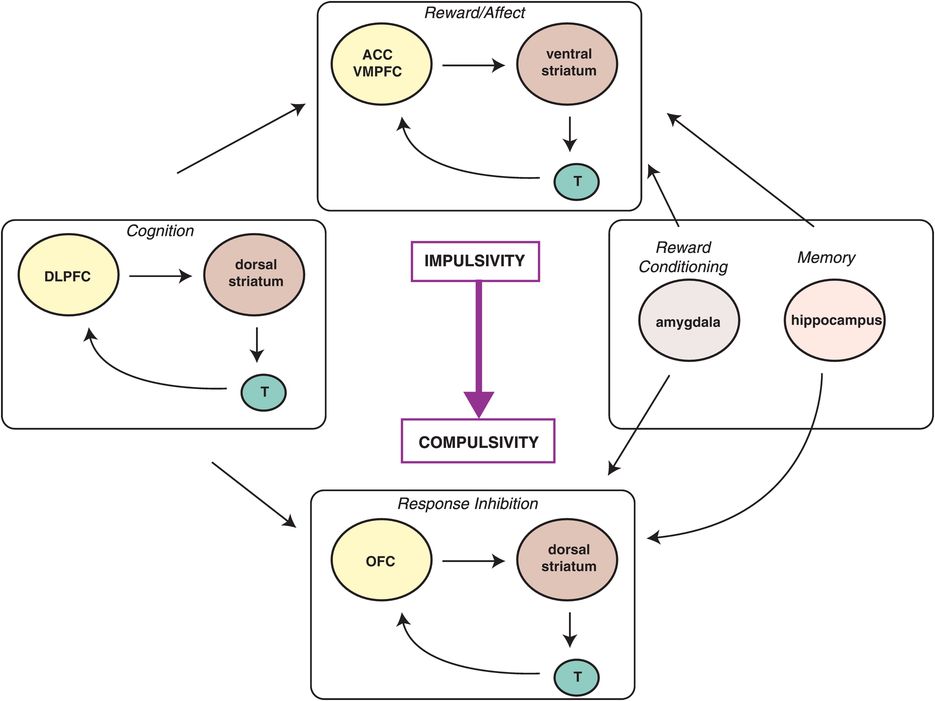
Figure 14-4. Spiraling circuits of impulsivity and compulsivity. The progression from occasional, impulsive drug use to compulsive use and addiction involves both the dysregulation of bottom-up reward circuits and insufficient top-down inhibition of these circuits. The amygdala and hippocampus provide regulatory input to this system as well. ACC, anterior cingulate cortex; DLPFC, dorsolateral prefrontal cortex; OFC, orbitofrontal cortex; T, thalamus; VMPFC, ventromedial prefrontal cortex.
A well-known example of ventral to dorsal migration is drug addiction. Although initial drug use is thought to be voluntary and linked to trait impulsivity, drug abusers gradually lose control over drug-seeking and drug-taking behavior, which becomes compulsive (Figure 14-5). Impulses to take drugs or to perform certain behaviors initially give a “high” or at least great pleasure and satisfaction (Figure 14-5). If this happens infrequently enough so as not to trigger neuroplastic cascades from ventral to dorsal, this remains under relative control, and can be seen as being occasionally “naughty” (Figure 14-5). However, impulsive drug use or impulsive behaviors repeated too often may progress to compulsive use driven by a desire to reduce the distressing symptoms of withdrawal that develop over time as the drug/behavior is repeated many times (Figure 14-5). Individuals with drug or behavioral addictions experience tension and arousal in anticipation of performing the behavior but dysphoric mood (but no physiological withdrawal) when prevented from performing the behavior or taking the drug. The pleasure and gratification that the drug/behavior initially causes, however, diminish over time, perhaps requiring increasing doses (e.g., gambling higher dollar amounts, more amounts or frequency of drug ingestion) in order to achieve the same effects (akin to tolerance) (Figure 14-5).
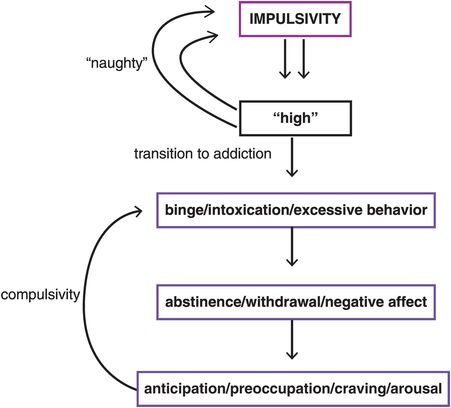
Figure 14-5. Shifting from impulsivity to compulsivity. Drug addiction provides a good example of the shift from impulsivity to compulsivity that comes with migration from ventral to dorsal circuits. The impulse to take a drug initially leads to great pleasure and satisfaction (a “high”). If this happens infrequently, the behavior may be a bit “naughty” but will not necessarily progress to compulsivity. With chronic substance use, compulsivity may develop as an individual’s drive turns from seeking pleasure to seeking relief from distressing symptoms of withdrawal and anticipation of obtaining the drug.
Maybe the first dose of any drug will always be the best one, most reinforcing and without penalty. However, individuals do not usually just take one dose of a drug, or perform satisfying behaviors just once. High impulsivity predisposes to the development of compulsions and is predictive of over-reliance on habit learning. Accelerated habit formation may underlie the transition in individuals who have high impulsivity to compulsions and habits. Compulsivity is clearly a maladaptive perseveration of behavior. This is not so much being naughty and giving into temptations (Figure 14-5) as being more like one of Pavlov’s dogs with a mindless involuntary conditioned compulsive response, with willpower being totally inadequate to interrupt the potentially destructive perseverations of behavior once they have become compulsive habits (Table 14-2 and Figure 14-5).
The mesolimbic dopamine circuit as the final common pathway of reward
All drugs that can lead to addiction increase dopamine (DA) in ventral striatum, also called the nucleus accumbens. This area of the brain is familiar to readers, as it is the same area discussed in Chapter 4 on psychosis and also known as the mesolimbic dopamine pathway hypothesized to be overly active in psychosis and to mediate the positive symptoms of schizophrenia (see Figures 4-12, 4-13, 4-30, 4-31). The final common pathway of reinforcement and reward in the brain is also hypothesized to be this same mesolimbic dopamine pathway (Figure 14-6). Some even consider this to be the “center of hedonic pleasure” of the brain and dopamine to be the “neurotransmitter of hedonic pleasure.” There are many natural ways to trigger your mesolimbic dopamine neurons to release dopamine, ranging from intellectual accomplishments to athletic accomplishments, to enjoying a good symphony, to experiencing an orgasm. These are sometimes called “natural highs” (Figure 14-6). The inputs to the mesolimbic pathway that mediate these natural highs include a most incredible “pharmacy” of naturally occurring substances ranging from the brain’s own morphine/heroin (endorphins), to the brain’s own marijuana (anandamide), to the brain’s own nicotine (acetylcholine), to the brain’s own cocaine and amphetamine (dopamine itself) (Figure 14-7).
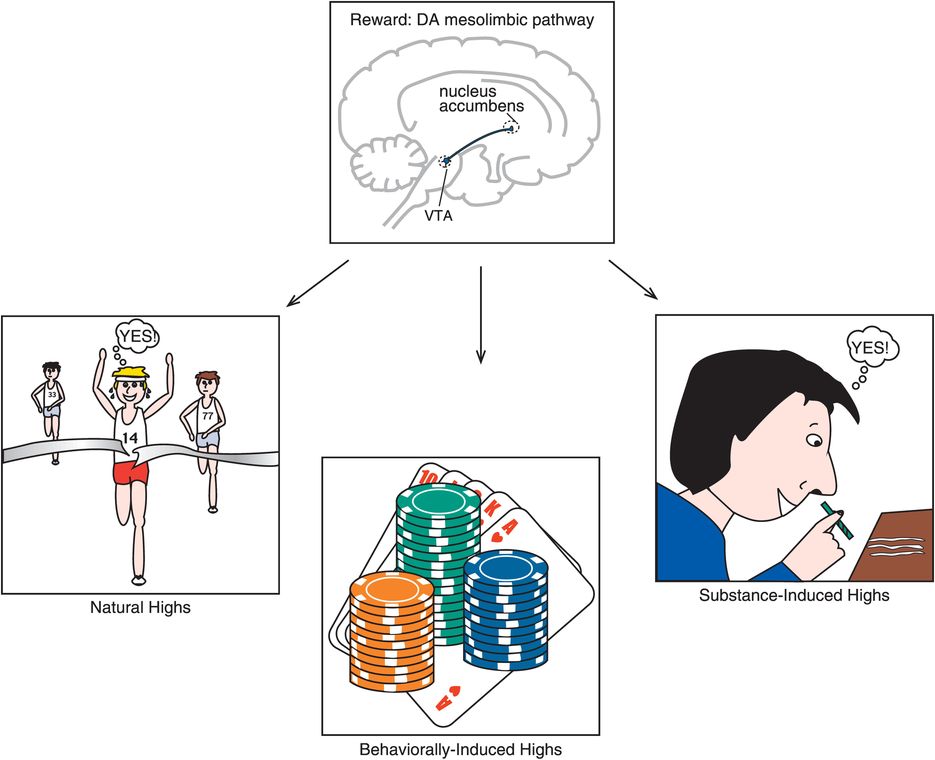
Figure 14-6. Dopamine is central to reward. Dopamine (DA) has long been recognized as a major player in the regulation of reinforcement and reward. Specifically, the mesolimbic pathway from the ventral tegmental area (VTA) to the nucleus accumbens seems to be crucial for reward. Naturally rewarding activities, such as achieving major accomplishments or enjoying a good meal, can cause fast and robust increases in DA in the mesolimbic pathway. Drugs of abuse also cause DA release in the mesolimbic pathway. In fact, drugs of abuse can often increase dopamine in a manner that is more explosive and pleasurable than that which occurs naturally. Unfortunately, unlike a natural high, the activation caused by drugs of abuse can eventually cause changes in reward circuitry that are associated with a vicious cycle of drug preoccupation, craving, addiction, dependence, and withdrawal. This conceptualization has similarities to many impulsive–compulsive disorders such as pathological gambling. That is, individuals with these disorders experience tension and arousal in anticipation of performing the behavior and dysphoric mood (but no physiological withdrawal) when prevented from performing the behavior. In addition, the pleasure and gratification that is initially experienced when performing the behavior seems to diminish over time, perhaps requiring increasing “doses” (e.g., gambling higher dollar amounts) in order to achieve the same effects (akin to tolerance).
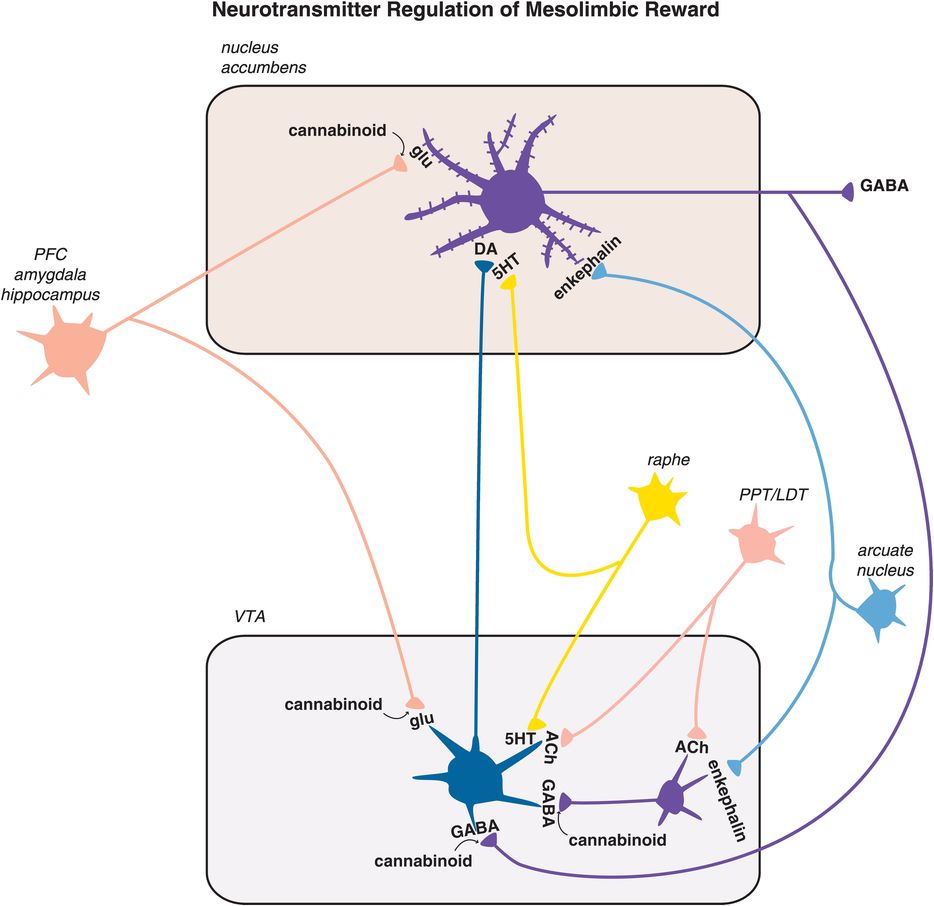
Figure 14-7. Neurotransmitter regulation of mesolimbic reward. The final common pathway of reward in the brain is hypothesized to be the mesolimbic dopamine pathway. This pathway is modulated by many naturally occurring substances in the brain in order to deliver normal reinforcement to adaptive behaviors (such as eating, drinking, sex) and thus to produce “natural highs,” such as feelings of joy or accomplishment. These neurotransmitter inputs to the reward system include the brain’s own morphine/heroin (i.e., endorphins such as enkephalin), the brain’s own cannabis/marijuana (i.e., anandamide), the brain’s own nicotine (i.e., acetylcholine), and the brain’s own cocaine/amphetamine (i.e., dopamine itself), among others. The numerous psychotropic drugs of abuse that occur in nature bypass the brain’s own neurotransmitters and directly stimulate the brain’s receptors in the reward system, causing dopamine release and a consequent “artificial high.” Thus alcohol, opioids, stimulants, marijuana, benzodiazepines, sedative hypnotics, hallucinogens, and nicotine all affect this mesolimbic dopaminergic system. PFC, prefrontal cortex; PPT/LDT, pedunculopontine tegmental and laterodorsal tegmental nuclei.
The numerous psychotropic drugs of abuse also have a final common pathway of causing the mesolimbic pathway to release dopamine, often in a manner more explosive and pleasurable than that which occurs naturally. Also, it now appears that potentially maladaptive behaviors as well as drugs can result in the release of dopamine that in turn stimulates the reward system (Figure 14-7). These are included in the impulsive–compulsive disorder construct (Figure 14-1) and include behaviors such as gambling, using the internet, shopping, and even eating. Drugs bypass the brain’s own neurotransmitters and directly stimulate the brain’s own receptors for these drugs, causing dopamine to be released. Since the brain already uses neurotransmitters that resemble drugs of abuse, it is not necessary to earn your reward naturally since you can get a much more intense reward in the short run and upon demand from a drug of abuse than you can from a natural high with the brain’s natural system. However, unlike a natural high, a drug-induced reward can start a cascade of neuroadaptation in the ventral striatum loop that migrates to the dorsal striatal loop (Figure 14-1), such that the initial high caused by impulsive early use of a drug leads to withdrawal, craving, and preoccupation with finding drug, thus beginning a vicious cycle of abuse, addiction, dependence, and withdrawal (Figure 14-5).
Substance addictions
Not everyone who takes a drug once gets addicted to it. Why? For one thing, some drugs seem to be intrinsically more addicting than others (Table 14-3). For another, some individuals may be more impulsive by nature or have a genetically dysfunctional reward system. It seems that impulsive traits and a dysfunctional reward system may confer a propensity towards drug use and abuse, and when drugs are ingested frequently, impulsive drug use can recruit the involvement of the habit system perhaps in some individuals more readily than in others, triggering neuroplasticity in the compulsivity circuit, which hypothetically is the means by which drug ingestion eventually becomes compulsive in some individuals (Figures 14-1 and 14-5).
| Probability of becoming dependent when you have tried a substance at least once | |
|---|---|
| Tobacco | 32% |
| Heroin | 23% |
| Cocaine | 17% |
| Alcohol | 15% |
| Stimulants | 11% |
| Anxiolytics | 9% |
| Cannabis | 9% |
| Analgesics | 8% |
| Inhalants | 4% |
Stimulants
The speed with which a drug enters the brain dictates the degree of the subjective “high” (Figure 14-8). This may be the reason why drugs that are inhaled, snorted, or injected, thus entering the brain in a sudden explosive manner, are usually much more reinforcing than when those same drugs are taken orally, where speed of entry to the brain is considerably slowed by the process of gastrointestinal absorption. Cocaine is not even active orally, so users have learned over the years to take it intranasally – where the drug rapidly enters the brain directly, bypassing the liver, and thus can have a more rapid onset this way than even with intravenous administration. The most rapid and robust way to deliver drugs to the brain is to smoke those that are compatible with this route of administration, as this avoids first-pass metabolism through the liver and is somewhat akin to giving the drug by intra-arterial/intra-carotid bolus via immediate absorption across the massive surface area of the lung. The faster the drug’s entry into the brain, the stronger are its reinforcing effects, probably because this form of drug delivery triggers phasic DA firing, the type associated with reward and saliency (see Chapter 12 for discussion, and Figures 12-10, 12-29, and 12-31).
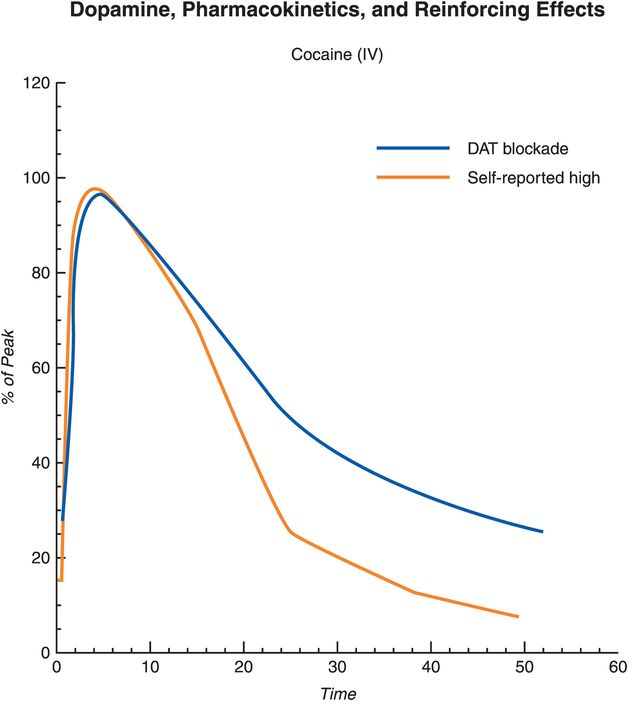
Figure 14-8. Dopamine, pharmacokinetics, and reinforcing effects. Acute drug use causes dopamine (DA) release in the striatum. However, the reinforcing effects of the drug are largely determined not only by the presence of DA but also by the rate at which DA increases in the brain, which in turn is dictated by the speed at which the drug enters and leaves the brain, in this case targeting the dopamine transporter (DAT). An abrupt and large increase in DA (such as that caused by drugs of abuse that block DAT) mimic the phasic DA firing associated with conveying information about reward and saliency. The rate of drug uptake is subject to the route of administration, with intravenous administration and inhalation producing the fastest drug uptake, followed by snorting. In addition, different drugs of abuse have different “reward values” (i.e., different rates at which they increase DA) based on their individual mechanisms of action.
On the other hand, some of these very same stimulants taken at low doses orally, especially within controlled-release formulations that minimize peak absorption, slow the rate of absorption, and prolong the duration of drug exposure, are not particularly reinforcing, but instead are therapeutic agents for treating ADHD, as discussed in Chapter 12 and illustrated in Figures 12-29 and 12-30. As discussed in Chapter 12, hypothetically, stimulants administered in this slow-release manner act to “tune” inefficient brain circuits by targeting the prefrontal cortex, enhancing tonic dopamine firing for motivation and attention, and reducing impulses and hyperactivity, while allowing sufficient phasic dopamine firing for learning and for facilitating appropriate goal-directed behaviors/reward (Figures 12-10 through 12-31). Although therapeutic actions of stimulants are thought to be directed at the prefrontal cortex to enhance both norepinephrine and dopamine neurotransmission there (Figures 12-13 through 12-18; Figure 12-31), the reinforcing effects and abuse of stimulants are thought to be directed at reward circuits, especially at dopamine release from mesolimbic dopamine neurons in the nucleus accumbens (Figure 12-31).
It turns out that in the long run it is not the reward of drug, but the anticipation of the reward, that is associated with drug seeking, or food seeking, or seeking numerous other types of situations involved in a wide range of impulsive–compulsive disorders (Table 14-1). Dopamine neurons actually stop responding to the primary reinforcer (i.e., the drug, the food, gambling) and instead begin to respond to the conditioned stimulus (i.e., the sight of the drug, the refrigerator door, the gambling casino). Conditioned responses underlie craving and compulsive use, and the increased dopamine migrates to the dorsal striatum (Figures 14-1 and 14-4). Drugs and behaviors may initially lead to dopamine increase in the ventral striatum and reward (Figures 14-1, 14-2, 14-4, 14-6, 14-7), but with repeated administration, as habits develop, dopamine increases shift from the drug/behavior to the conditioned response/environmental trigger, as the dopamine increases shift from the ventral striatum/nucleus accumbens (Figure 14-2) to the dorsal striatum (Figure 14-3).
Dopamine is associated with motivation, and the motivation to procure drugs is the hallmark of addiction. Drug seeking and drug taking become the main motivational drive when one is addicted, and thus the addicted subject is aroused and motivated when seeking to procure the drug, but is withdrawn and apathetic when exposed to non-drug-related activities (Figures 14-5 and 14-8). What starts out as increased DA release leading to increased ventral striatum and anterior cingluate cortex (ACC) activity with reward may end up as a compulsive drive with escalating dosing in an attempt to get increased reward stimulation to restore a resultant DA deficiency. The discrepancy between expectation for drug effects and the blunted DA effects maintains drug taking in an attempt to achieve the expected reward. High doses of stimulants can cause tremor, emotional lability, restlessness, irritability, panic, and repetitive stereotyped behavior. At even higher repetitive doses, stimulants can induce paranoia and hallucinations, with hypertension, tachycardia, ventricular irritability, hyperthermia, and respiratory depression. In overdose, stimulants can cause acute heart failure, stroke, and seizures.
Not only methylphenidate and amphetamines, but also cocaine are all inhibitors of the dopamine transporter (DAT) and the norepinephrine transporter (NET) (see discussion in Chapter 12, and Figures 12-25 and 12-28). Cocaine also inhibits the serotonin transporter (SERT) and is also a local anesthetic, which Freud himself exploited to help dull the pain of his cancer of the jaw and mouth. He may have also exploited the second property of the drug, which is to produce euphoria, reduce fatigue, and create a sense of mental acuity due to inhibition of dopamine reuptake at the dopamine transporter, at least for a while, until drug-induced reward is replaced by drug-induced compulsivity.
Although there are no approved treatments for stimulant addicts, in the future there may be a cocaine vaccine that removes the drug before it reaches the brain so there are no more reinforcing effects that accompany drug ingestion. Theoretically, it may also be possible to administer intravenously a long-acting form of the enzyme cocaine esterase that destroys cocaine before it can exert its reinforcing effects, as has been shown in animal models. Naltrexone, a µ-opioid antagonist approved for the treatment of both opioid and alcohol addiction, is also being investigated for patients with stimulant addiction, particularly for those patients with polydrug dependence on both the opioid heroin and the stimulant amphetamine. Buprenorphine, a synthetic opioid used for treatment of pain and for opioid addiction, stimulates as a partial agonist both µ- and κ-opioid receptors, and can decrease cocaine use in opioid addicts. It is also being studied in combination with naltrexone for cocaine addicts who do not have opioid addiction. The combination results in stimulation only of κ-opioid receptors and not of µ-opioid receptors and may decrease compulsive cocaine self-administration in animals without producing opioid addiction – suggesting that, at least in this case, three drugs might be better than one!
Nicotine
How common is smoking in clinical psychopharmacology practices? Some estimates are that more than half of all cigarettes are consumed by patients with a concurrent psychiatric disorder, and that smoking is the most common comorbidity among seriously mentally ill patients. It is estimated that about 20% of the general population (in the US) smoke, about 30% of people who regularly see general physicians smoke, but that 40–50% of patients in a psychopharmacology practice smoke, including 60–85% of patients with ADHD, schizophrenia, and bipolar disorder. Unfortunately, histories of current smoking are often not carefully taken nor recorded as one of the diagnoses for smokers in mental health practices, and only about 10% of smokers report being offered treatment proactively by psychopharmacologists and other clinicians.
Nicotine acts directly upon nicotinic cholinergic receptors in reward circuits (Figure 14-7). Cholinergic neurons and the neurotransmitter acetylcholine are discussed in Chapter 13 and illustrated in Figures 13-17 through 13-24. Nicotinic receptors are illustrated in Figures 13-20 through 13-22. There are two major subtypes of nicotinic receptors that are known to be present in the brain, the α4β2 subtype and the α7 subtype (discussed in Chapter 13 and illustrated in Figure 13-20). Nicotine’s actions in the ventral tegmental area are those that are theoretically linked to addiction, namely at α4β2-nicotinic postsynaptic receptors on dopamine neurons, leading to dopamine release in the nucleus accumbens, and at α7-nicotinic presynaptic receptors on glutamate neurons, which causes glutamate release, and in turn dopamine release in the nucleus accumbens (Figure 14-9). The release-promoting actions of presynaptic α7-nicotinic receptors on glutamate neurons are discussed in Chapter 13 and illustrated in Figure 13-20. Nicotine also appears to desensitize α4β2 postsynaptic receptors on inhibitory GABAergic interneurons in the VTA (Figure 14-9); this also leads to DA release in nucleus accumbens by disinhibiting dopaminergic mesolimbic neurons. Actions of nicotine on postsynaptic α7-nicotinic receptors in the prefrontal cortex may be linked to the pro-cognitive and mentally alerting actions of nicotine, but not to addictive actions.
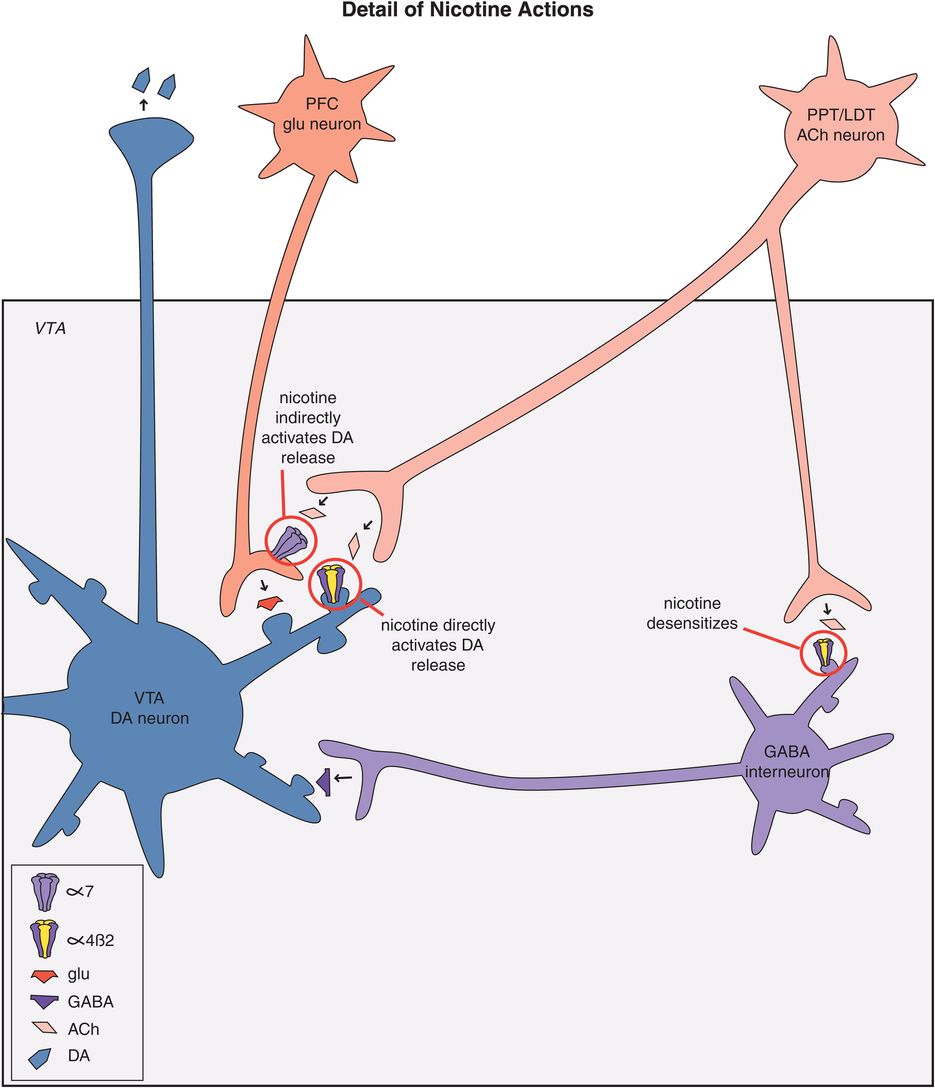
Figure 14-9. Actions of nicotine. Nicotine directly causes dopamine release in the nucleus accumbens by binding to α4β2-nicotinic postsynaptic receptors on dopamine neurons in the ventral tegmental area (VTA). In addition, nicotine binds to α7-nicotinic presynaptic receptors on glutamate neurons in the VTA, which in turn leads to dopamine release in the nucleus accumbens. Nicotine also seems to desensitize α4β2 postsynaptic receptors on GABA interneurons in the VTA; the reduction of GABA neurotransmission disinhibits mesolimbic dopamine neurons and thus is a third mechanism for enhancing dopamine release in the nucleus accumbens. PFC, prefrontal cortex; PPT/LDT, pedunculopontine tegmental and laterodorsal tegmental nuclei.
The α4β2-nicotinic receptors adapt to the chronic intermittent pulsatile delivery of nicotine in a way that leads to addiction (Figure 14-10). That is, initially these receptors in the resting state are opened by delivery of nicotine, which in turns leads to dopamine release and reinforcement, pleasure, and reward (Figure 14-10A). By the time the cigarette is finished, these receptors become desensitized, so that they cannot function temporarily and thus cannot react either to acetylcholine or to nicotine (Figure 14-10A). In terms of obtaining any further reward, you might as well stop smoking at this point. An interesting question to ask is: how long does it take for the nicotinic receptors to desensitize? The answer seems to be: about as long as it takes to inhale all the puffs of a standard cigarette and burn it down to a butt. Thus, it is probably not an accident that cigarettes are the length that they are. Shorter does not maximize the pleasure. Longer is a waste since by then the receptors are all desensitized anyway (Figure 14-10A).
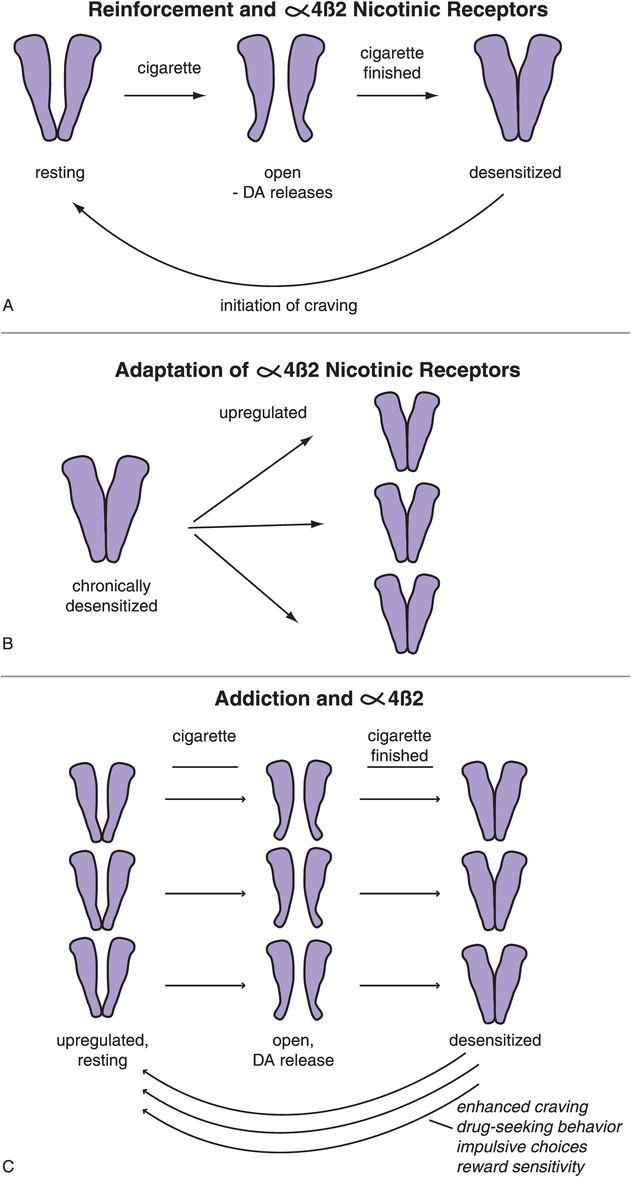
Figure 14-10. Reinforcement and α4β2 nicotinic receptors. (A) In the resting state α4β2 nicotinic receptors are closed (left). Nicotine administration, as by smoking a cigarette, causes the receptor to open, which in turn leads to dopamine release (middle). Long-term stimulation of these receptors leads to their desensitization, such that they temporarily can no longer react to nicotine (or to acetylcholine); this occurs in approximately the same length of time it takes to finish a single cigarette (right). As the receptors resensitize, they initiate craving and withdrawal due to the lack of release of further dopamine. (B) With chronic desensitization, α4β2 receptors upregulate to compensate. (C) If one continues smoking, however, the repeated administration of nicotine continues to lead to desensitization of all of these α4β2 receptors, and thus the upregulation does no good. In fact, the upregulation can lead to amplified craving as the extra receptors resensitize to their resting state.
The problem for the smoker is that when the receptors resensitize to their resting state, this initiates craving and withdrawal due to the lack of release of further dopamine (Figure 14-10A). Another interesting question is: how long does it take to resensitize nicotinic receptors? The answer seems to be: about the length of time that smokers take between cigarettes. For the average one pack per day smoker awake for 16 hours, that would be about 45 minutes, possibly explaining why there are 20 cigarettes in a pack (i.e., enough for an average smoker to keep his or her nicotinic receptors completely desensitized all day long).
Putting nicotinic receptors out of business by desensitizing them causes neurons to attempt to overcome this lack of functioning receptors by upregulating the number of receptors (Figure 14-10B). That, however, is futile, since nicotine just desensitizes all of them the next time a cigarette is smoked (Figure 14-10C). Furthermore, this upregulation is self-defeating because it serves to amplify the craving that occurs when the extra receptors are resensitizing to their resting state (Figure 14-10C).
From a receptor point of view, the goal of smoking is to desensitize all nicotinic α4β2 receptors, get the maximum dopamine release at first, but eventually mostly to prevent craving. Positron emission tomography (PET) scans of α4β2-nicotinic receptors in human smokers confirm that nicotinic receptors are exposed to just about enough nicotine for just about long enough from each cigarette to accomplish this. Craving seems to be initiated at the first sign of nicotinic receptor resensitization. Thus, the bad thing about receptor resensitization is craving. The good thing from a smoker’s point of view is that as the receptors resensitize, they are available to release more dopamine and cause pleasure or suppress craving and withdrawal again.
Treating nicotine dependence is not easy. There is evidence that nicotine addiction begins with the first cigarette, with the first dose showing signs of lasting a month in experimental animals (e.g., activation of the anterior cingulate cortex for this long after a single dose). Craving begins within a month of repeated administration. Perhaps even more troublesome is the finding that the “diabolical learning” that occurs from substance abuse of all sorts including nicotine may be very, very long-lasting once exposure to nicotine is stopped. Some evidence suggests that these changes even last a lifetime, with a form of “molecular memory” to nicotine, even in long-term abstinent former smokers. One of the first successful agents proven to be effective is nicotine itself, but in a route of administration other than smoking: gums, lozenges, nasal sprays, inhalers, and transdermal patches. Delivering nicotine by these other routes does not attain the high levels or the pulsatile blasts that are delivered to the brain by smoking, so they are not very reinforcing. However, these alternative forms of nicotine delivery can help to reduce craving due to a steady amount of nicotine that is delivered, presumably desensitizing an important number of resensitizing and craving nicotinic receptors.
Another treatment for nicotine dependence is varenicline, a selective α4β2-nicotinic acetylcholine receptor partial agonist (Figures 14-11 and 14-12). Figure 14-11 contrasts the effects of nicotinic partial agonists (NPAs) with nicotinic full agonists and nicotinic antagonists on the cation channel associated with nicotinic cholinergic receptors. Nicotinic full agonists include acetylcholine, a short-acting full agonist, and nicotine, a long-acting full agonist. They open the channel fully and frequently (Figure 14-11, left). By contrast, nicotinic antagonists stabilize the channel in the closed state, but do not desensitize these receptors (Figure 14-11, right). NPAs stabilize nicotinic receptors in an intermediate state which is not desensitized and where the channel is open less frequently than with a full agonist, but more frequently than with an antagonist (Figure 14-11, middle).
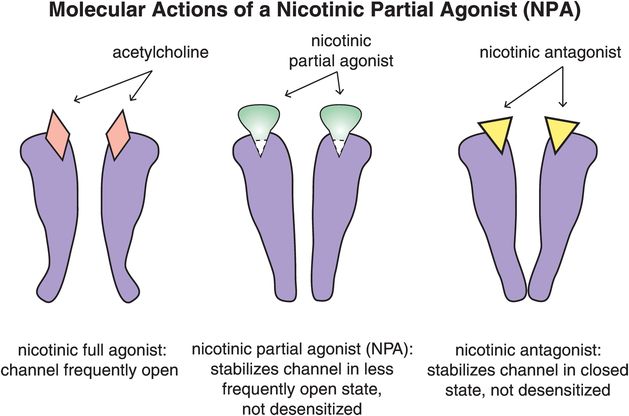
Figure 14-11. Molecular actions of a nicotinic partial agonist (NPA). Full agonists at α4β2 receptors, such as acetylcholine and nicotine, cause the channels to open frequently (left). In contrast, antagonists at these receptors stabilize them in a closed state, such that they do not become desensitized (right). Nicotinic partial agonists (NPAs) stabilize the channels in an intermediate state, causing them to open less frequently than a full agonist but more frequently than an antagonist (middle).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


