Fig. 8.1
VCAM-1 immunohistochemical staining on aorta. Normotensive rodent exhibited a relatively weaker staining on endothelial lining (EL; left) compared to hypertensive rodent, which showed a remarkably intense staining for VCAM-1 (right). Abbreviations: L lumen, TM tunica media. (Magnification: ×400; scale bar: 50 μm)
Intercellular Adhesion Molecule-1
Intercellular cell adhesion molecule-1 (ICAM-1 ) is a cell surface glycoprotein expressed on multiple cell types including leukocytes, epithelial cells, endothelial cells, and fibroblasts [25,26]. It is expressed constitutively and abundantly on vascular endothelial cells. Its expression is markedly induced by some pro-inflammatory stimuli like IL-1, TNF-α, interferon-γ (IFN), or LPS [27,28]. Structurally, ICAM-1 is related to the immunoglobulin superfamily of adhesion molecules including VCAM-1, which share a common immunoglobulin-like structure and comprise various proteins fundamentally function in both cell-to-cell and cell-to-matrix adherence [29,30]. ICAM-1 contains five extracellular immunoglobulin-like repeat, one transmembrane segment, and a short cytoplasmic tail [30]. A motif within the first and third domains is responsible for binding to the β2 integrin ligands such as lymphocyte function-associated antigen-1 (LFA-1) expressed on T-lymphocytes and Mac-1 on neutrophil, respectively [31,32]. Through these interactions, ICAM-1 alongside with VCAM-1 plays an important part in the recruitment and trafficking of leukocytes in endothelial cells during inflammatory responses, such as atherosclerosis [33]. While VCAM-1 is crucial during the initial step of leukocytes tethering via VLA-4 ligands, ICAM-1 functions to adhere the leukocytes firmly to the endothelial surface and initiate the process of immune cell transmigratory egress from the circulatory system [33]. Both VCAM-1 and ICAM-1 function to stabilise leukocyte adhesion to the endothelial lining. Endothelial cells, smooth muscle cells, and macrophages are the general sources of VCAM-1 and ICAM-1 expression in atherosclerotic lesions [34,35]. A study demonstrated that VCAM-1, but not ICAM-1, has a considerable role in the formation of early atherosclerotic lesions [36]. However, ICAM-1 was found to mainly involve in the progression of atherosclerosis [37]. Several early studies have reported that the upregulation of VCAM-1 and ICAM-1 is associated with the sites as well as stages of atherosclerotic lesion formation [38–40].
Recruitment of leukocytes to endothelial linings causes damage to the vascular wall. It has been shown recently that ICAM-1 expression was increased significantly in hypertensive rodents ([23]; Fig. 8.2) and in pregnancy-induced hypertensive women [41]. Elevated endothelial expressions of adhesion molecules may indicate a disturbance to the endothelial function [42]. A recent study has reported significant positive correlations of systolic and diastolic blood pressure with ICAM-1 and VCAM-1 and that a decrease in soluble adhesion molecules might suggest the deactivation of endothelium [43]. Therefore, hypertension is considered as a state of endothelial activation and dysfunction. Both VCAM-1 and ICAM-1 are useful markers for investigating vascular inflammation in hypertension.
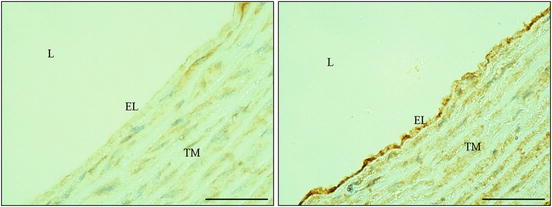
Fig. 8.2
ICAM-1 immunohistochemical staining on aorta. The immunostaining is denser in hypertensive rodent (right) than normotensive rodent (left). Abbreviations: EL endothelial lining, L lumen, TM tunica media. (Magnification: ×400; scale bar: 50 μm)
Lectin-like Oxidised Low-Density Lipoprotein Receptor-1
Lectin-like oxidised low-density lipoprotein receptor-1 (LOX-1 ) , a type II membrane protein (52 kD) with a C-terminus lectin-like extracellular domain and a short N-terminus cytoplasmic tail, is a relatively newly discovered receptor. This novel receptor was identified in vascular bovine aortic endothelial cells only a decade ago (Sawamura et al. 1997). It was identified as a major receptor for oxidised low-density lipoprotein (Ox-LDL) on endothelial cells. The C-terminal end residues and several conserved positively charged residues spanning the lectin domain are recognised for Ox-LDL binding [44,45]. It binds, internalises, and degrades the oxidatively modified Ox-LDL, but not native LDL [46]. Therefore, LOX-1 might be an essential mediator for the pathophysiological effects of Ox-LDL in the vascular wall.
Since its discovery, a tremendous number of studies have demonstrated a clearer definition and understanding of the central role played by LOX-1 in atherosclerosis [47–52]. Endothelial dysfunction elicited by Ox-LDL is recognised as the key step in the initiation of atherosclerosis. LOX-1 activation by Ox-LDL causes endothelial functional alterations that are characterised by induction of adhesion molecules [53], monocyte-chemotactic protein [54], smooth muscle cells’ proliferation [55], and attenuated endothelium-dependent vasodilatory response by reducing the generation of nitric oxide [56]. Hence, LOX-1 is unquestionably a confirmative marker for atherosclerosis.
Unexpectedly, however, LOX-1 expression was remarkably upregulated (>20 folds) in the aorta of hypertensive rats, which implies a pathophysiologic role for LOX-1 in hypertension or in hypertensive vascular remodelling [57]. Furthermore, the LOX-1 upregulation in the kidney of Dahl salt-sensitive hypertensive rats was parallel to renal dysfunction, suggesting a possible role of LOX-1 in hypertensive renal disease [58]. We have also found a slight increase in the expression of aortic endothelial LOX-1 in hypertensive rats ([23], Fig. 8.3). LOX-1 may be involved in impairing the endothelium-dependent vasodilatation in these experimental animals. Nonetheless, the LOX-1 expression was found to show a significant direct association with the expressions of VCAM-1 and ICAM-1 in hypertensive group. Inoue et al. [59] have reported that LOX-1 overexpression promoted the expression of VCAM-1 and ICAM-1 on the endothelial cells, which further contributes to vascular inflammation. Hence, it is obvious that LOX-1 could be a novel marker for hypertension. The pathogenetic roles of LOX-1 in hypertension development, however, remain to be determined. Investigating the functions of LOX-1 in hypertension development may represent a novel therapeutic target to control the disease.
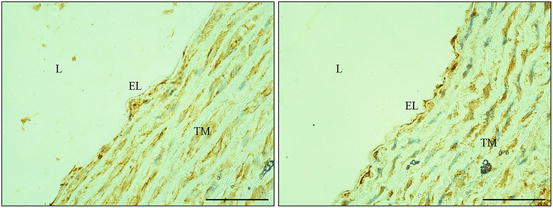
Fig. 8.3
LOX-1 immunohistochemical staining on aorta. Hypertensive rodent demonstrated a relatively stronger staining on endothelial lining (EL; right). On the contrary, little or absence of staining on EL in normotensive rodent (left). Abbreviations: L lumen, TM tunica media. (Magnification: ×400; scale bar: 50 μm)
P-selectin
The selectin family of adhesion molecules mediates the initial attachment of leukocytes to endothelial surface before their firm adhesion at inflammatory sites. The selectin family consists of three closely associated members sharing common structural features but with differential expression by leukocytes (L-selectin), platelets (P-selectin), and vascular endothelium (P selectin and E-selectin). Among these molecules, P-selectin is of interest because of its unique expression by both platelets and endothelial cells. It is the largest of selectin family with a mass of approximately 140 kDa. P-selectin is also previously known as granule membrane protein 140 (GMP-140) or platelet activation-dependent granule external membrane protein (PADGEM) [60]. It is a cell membrane component of the α granules of platelets (that is how it gets its name) as well as of the Weibel–Palade body (an organelle that stores the coagulation-related von Willebrand factor in endothelial cells). P-selectin is re-localised rapidly from the cytoplasmic bodies to the cell surface upon stimulation by inflammatory mediators [61] as its expression has been observed on the endothelial lining of blood vessel by immunohistochemistry [62].
Structurally, P-selectin consists of an extracellular N-terminus lectin domain, an epidermal growth factor-like region, nine complement regulatory-like protein repeats, a transmembrane section, and a short intracytoplasmic tail [63]. P-selectin binds to its specific mucin-like counter-receptor, designated as P-selectin glycoprotein ligand-1 (PSGL-1) on leukocytes [64], suggesting that it is involved in an interaction between leukocytes and the endothelium, between platelets and leukocytes, and also between platelets and the endothelium themselves [65–67].
Several studies have highlighted the essential role of P-selectin in tethering leukocytes on the endothelial surface and the interaction between platelets and leukocytes [68–70]. Therefore, raised plasma level of P-selectin is considered to be a marker of inflammation. Various experiments have reported raised P-selectin expression in cardiovascular diseases such as stable peripheral artery disease [71], atherosclerosis [72], and unstable angina [73]. Immunohistochemical analysis of human atherosclerotic plaques has demonstrated a strong expression of P-selectin by the endothelial cells overlying active atherosclerotic plaques and fatty streaks [74]. Nevertheless, a study demonstrated a significant and positive correlation of blood pressure and P-selectin that persists in the presence of extreme blood pressure elevation [75]. Increased blood pressure was suggested to be related to an elevated level of plasma P-selectin and endothelial dysfunction (as indicated by high von Willebrand factor levels) [76]. Quite recently, inhibition of P-selection function by inclacumab has been shown to diminish the platelet–leukocyte interaction which may represent an effective treatment for cardiovascular diseases [77]. As it may be involved in and link up various cardiovascular diseases, the expression of P-selectin in hypertension therefore deserves additional studies.
E-selectin
Generally, P-selectin and E-selectin are important in leukocytes recruitment. Both of them are induced at reasonably high density on the vascular endothelial cells at sites of inflammation. Halacheva et al. [78] observed that positive immunostaining for E-selectin on arterial endothelium was accompanied by infiltration of inflammatory cells in the intima. Due to their overlapping function in leukocyte adhesion, P-selectin seems to outshine E-selectin as mice lacking this molecule only exhibit a modest reduction in the atherosclerotic lesion size [79,80]. However, a study has reported the combined effect of these selectins on the development of atherosclerotic lesion [81]. In that study, P- and E-selectin-double-deficient mice were found to develop fatty streaks that were three times smaller than those developed in P-selectin-single-deficient mice.
Unlike ICAM-1 and VCAM-1, E-selectin is expressed only on the endothelial surface after pro-inflammatory stimulation and it can hence be considered a specific marker of endothelial dysfunction in cardiovascular and inflammatory diseases, including hypertension. The imperative involvement of E-selectin in hypertension has been increasingly ascertained by multiple lines of studies [18,82,83]. The possible mechanism played by E-selectin in raising blood pressure may be associated with impaired vasodilatory response and reduced endothelial nitric oxide production [84]. Nonetheless, E-selectin might as well be involved in vascular wall hypertrophy observed in hypertensive animals [85] because it has been previously found to be directly correlated to intima-media thickness in patients with ischemic heart disease [86]. The potential function of E-selectin in hypertension needs to be further elucidated.
Endothelin Receptors
The endothelin system consists of potent endogenous vasoconstrictor peptides that play pivotal roles in vasomotion, vascular smooth muscle cell proliferation, and vascular remodelling. Disturbance in its productions may contribute to elevated peripheral resistance in pathogenesis of hypertension [87]. Endothelin binds to a specific receptor on vascular smooth muscle cells to induce vasoconstriction. Endothelin receptors are classified as subtypes A, B, and C (ETA, ETB, and ETC), respectively [88]. While the knowledge on the subtype C receptor is limited, the ETA and ETB receptors have been extensively studied. The ETA receptors are expressed on vascular smooth muscle cells and function in vasoconstriction and proliferation [89]. Whereby the ETB receptors predominate on endothelial cells and are present in low densities in vascular smooth muscle cells of aorta, coronary arteries, mesenteric arteries, and veins of laboratory animals and in humans [90]. The ETB receptors mediate vasodilatation of smooth muscle via the production of nitric oxide and prostacyclin (PGI2) [91]. Due to its vasodilatory role, the ETB receptors have been receiving tremendous attentions lately as it may shed light on a potential alternative treatment for diseases involving endothelin disorder [92–94]. Interestingly, the function of ETB receptors may switch from vasodilatation mode (vasorelaxing phenotype, termed as ETB1) to vasoconstriction mode (contractile phenotype, termed ETB2) under certain pathophysiological conditions [95]. For instance, inflammatory cytokine TNF-α has been recently found to stimulate the vascular smooth muscle cell expression of the contractile ETB2 receptors in rodents via activating NF-κB pathway [96]. Immunohistochemical staining on paraffin-embedded arterial sections demonstrated upregulation of ETB2 receptors in tunica media after TNF-α intervention [96]. The novel function of these endothelin receptors represents an interesting study in hypertension.
Vascular Endothelial Growth Factor
Vascular endothelial growth factor (VEGF) is a potent endothelial cell mitogen and fundamental regulator of physiological and pathological angiogenesis. In rodent’s circulatory system, VEGF expression is demonstrated by immunohistochemistry on cardiac myocytes, aortic endothelium, and the tunica media of inferior vena cava [97]. VEGF regulates endothelial cell proliferation and migration [98]. Furthermore, it has been shown to induce endothelium-dependent vasodilation, leading to hypotensive effect [99]. VEGF signalling is mediated through interaction with two transmembrane endothelial cell receptor tyrosine kinases: VEGF receptors (VEGFR) types 1 (Flt-1) or VEGFR 2 (KDR). Binding of VEGF to its receptors causes intracellular phosphorylation such as of endothelial nitric oxide synthase (eNOS) resulting in an increased production of nitric oxide, which acts directly on endothelial cells and eventually leads to endothelial cell proliferation, permeability, and blood vessel formation [100]. A study has demonstrated that although the vasodilation and hypotensive effect of VEGF involves both receptors, the KDR is the major receptor mediating this effect [101]. Blood pressure-lowering effect of VEGF has been shown to be accompanied by upregulation of nitric oxide level [102]. Inhibiting the VEGF pathway may lead to hypertension [103]. However, there are studies reporting elevated VEGF expression in both hypertensive human and experimental rats [104–106]. It has been suggested that aside from its central role in angiogenesis, VEGF may also be involved in inflammatory processes of the vascular endothelial cells likely attributable to increased vascular permeability [107]. VEGF may possess multifaceted roles [108]. Investigations into the role played by VEGF in hypertension may lead to exciting findings .
Tumour Necrosis Factor Alpha
Tumour necrosis factor alpha (TNF-α) is another vascular inflammation marker that may be applicable for immunohistochemistry to study the relationship between inflammation and hypertension. It is a pro-inflammatory glycoprotein peptide hormone that plays a significant role in the initial activation of the immunological responses. Recent data have demonstrated the association between an elevated expression of TNF-α and raised blood pressure as well as cardiac dysfunction [109–111], suggesting an important pro-hypertensive role of inflammatory process in the vascular wall. Furthermore, blockage of TNF-α has been shown by immunohistochemical approach to reduce endothelial and smooth muscle cell TNF-α protein expression, which subsequently improves haemodynamic properties and attenuates inflammation in rodents [112]. The exact mechanisms by which TNF-α influences hypertension are still uncertain, but TNF-α has been reported to downregulate endothelial nitric oxide synthase (eNOS) mRNA expression [113], hence causing an inverse relationship between TNF-α and nitric oxide serum levels in rats [111]. Vascular inflammation induced by TNF-α is associated with impaired endothelium-dependent vasodilatory response [114].
Inducible Nitric Oxide Synthase
Nitric oxide is an important endogenous vasodilator in cardiovascular system that regulates systemic and local blood flow. It is synthesised by the catalytic action of a small family of enzymes known as nitric oxide synthase (NOS) , comprising of three isoforms: neuronal (nNOS), endothelial (eNOS), and inducible NOS (iNOS) [115]. In normal cardiovascular function, nitric oxide is produced predominantly by eNOS to play a central role in preserving physiological endothelial functions. However, in pathological conditions such as inflammation, iNOS, which is expressed mostly in smooth muscle cells, is stimulated to produce nitric oxide a thousand times more than that produced by eNOS [116,117]. While the relatively low amounts of nitric oxide by eNOS function to protect the cardiovascular system, this exaggerated release of nitric oxide via iNOS has been linked to the production of deleterious oxidative products [118]. Excessive amounts of oxidative products cause detrimental cardiovascular effects, which may be attributable to the reaction between superoxide and nitric oxide-forming peroxynitrite, subsequently promoting the so-called nitrosative stress and endothelial dysfunction [119,120]. A great number of recent reports have pointed out the significant role of iNOS expression in decreased nitric oxide bioavailability, increased oxidative stress, cell damage, impaired endothelial function, inflammatory activation, and eventually the development of hypertension [115,118,121–123]. Moreover, a polymorphism in iNOS gene which is associated with predisposition to hypertension and responsiveness to antihypertensive therapy has been identified in human subjects recently [124], further showing the important role of derangement in nitric oxide in the pathophysiology of hypertension. Hence, iNOS is a very useful marker for hypertension, inflammation, and oxidative stress .
Angiotensin II and Its Receptor
Angiotensin II (Ang II ) in hypertension is imperative because of its pluripotent properties. Ang II is the major bioactive component of the renin-angiotensin system produced locally in several tissues, including vascular wall, with a central role in the regulation of blood pressure, vasomotion, and sodium and water balance [125,126]. Furthermore, a recent study has reported that Ang II-induced hypertension involves the upregulation of lipogenic gene expression and dysfunction of both the systolic and diastolic cardiac performance in rats [127]. Aside from its involvement in blood pressure regulation, several studies have revealed the pro-inflammatory-inducing effects of Ang II in vascular endothelial cells [128,129]. Inhibition of angiotensin-converting enzyme (ACE) has been shown to reduce the Ang II-induced expressions of inflammatory cytokines [130,131]. In addition, Ang II also increases the generation of intracellular reactive oxygen species in endothelial cells, leading to excessive oxidative stress that may impair endothelial function [132]. This pro-oxidative effect of Ang II is conferred through the Ang II type I (AT1) receptor-nicotinamide adenine dinucleotide phosphate-oxidase (NADPH) pathway induction of reactive oxygen species production [133–135]. AT1 receptors are absolutely required by Ang II in the development of hypertension and cardiac hypertrophy [136]. Due to their adjacent expressions, immunohistochemistry may be a useful method to study the local effects of Ang II and AT1 receptor .
Principles of Immunohistochemistry
Considering the increasingly strong footing of the role of vascular wall inflammation in hypertension, immunohistochemistry has become an important method to investigate the inflammatory responses in circulatory system. Immunohistochemistry is a method used to recognise the presence and location of constituents of tissues (as antigens) in situ by means of corresponding labelled antibodies. The principle of immunohistochemistry lies on the immunoreaction between antibodies to bind specifically to their corresponding antigens. Since antibodies are specific, they will only bind to the antigens of interest in the tissue. The resulting reaction is visualised by the antibodies which are pre-labelled either with a substance that absorbs or emits light (fluorescent detection) or a substance that produces distinct colour (chromogenic detection). Therefore, immunohistochemistry is actually a general term referring to a group of immunostaining techniques including immunofluorescence techniques and immunoperoxidase techniques, in which specially labelled antibodies are applied for detecting proteins of interest.
Although immunohistochemistry is quantitatively less sensitive than other immunoassays such as protein Western blotting or enzyme-linked immunosorbent assay (ELISA), it however gives an overview of an intact tissue in the aspect of the localisation of a particular protein (i.e. in nucleus, cellular membrane, or cytoplasm). Since it is done on a certain tissue rather than the measuring of inflammatory biomarker levels in serum by other methods, say ELISA, immunohistochemistry is like “talking to the cells” and thus the expressions of inflammatory biomarkers may indicate what they are doing in vivo. The results are most likely reflecting what is going on in the vascular wall itself. It provides information about the presence and activation of a protein in a tissue, and it therefore helps to make sense of the numerical data obtained from other methods. Furthermore, due to its wide availability, lower cost, and easy and long preservation of the stained slides, immunohistochemistry has become an important method in the assessment of pathologies [137]. Only a small amount of tissue is needed to detect various parameters in immunohistochemistry. In addition, the continuing advance in computer-aided image analysis has shifted immunohistochemistry from a traditional “merely descriptive” technique to a quantitative measurement. Digital image evaluation has enabled measurements of protein activations by quantifying the colour density of positive immunostaining on a tissue. Therefore, immunohistochemistry is advantageous in providing not only qualitative but also semi-quantitative or quantitative information of protein expressions. Immunohistochemistry selectively detects specific proteins in tissue and has become a routine technique for diagnostic and assessment purposes of certain diseases. Moreover, it is widely applied for experimental purposes to unravel the pathophysiologic mechanisms.
Technical Aspects
Scrupulous attention to the technical details and components when conducting immunohistochemical staining promises easily interpretable slides. From tissue fixation, to antigen retrieval, to staining procedure, every aspect is crucial in determining the outcome.
Specimen Handling
In the circulatory system o f a rodent, aside from the heart, aorta is probably a more suitable tissue to be used in studying vascular pathologies due to its larger size and easier excision. From aortic arch, to thoracic aorta, to abdominal aorta, it can be used in immunohistochemical procedure [23,138–140]. Of course, other tissues from the circulatory system such as penile artery [141] or cerebral blood vessel [142] are applicable as well, depending on the aim of an experiment. The very first step in tissue handling is paramount in governing the quality of immunohistochemical results. Necrotic degradation starts immediately once the tissue is separated from its source of nutrients. Hence vascular specimen must be processed properly and rapidly isolated from adherent adipose and connective tissues after harvesting from the animal. Specimen should then be trimmed and cut for fixation. Size of specimen is important. For instance, aorta should be cut into short length so that the fixative can penetrate efficaciously into the tissue. The shorter or thinner the tissue, the faster the fixation can occur. Besides, fixative should be made sure that it is always fresh. After fixation, the specimen is paraffin embedded either longitudinally or horizontally. To study the expressions of vascular wall proteins, longitudinal embedding is recommended. Horizontal embedding is usually done to investigate the entire length of elastic lamellae or vascular smooth muscle cells. Extra care must be taken when longitudinally embedding a blood vessel sample to ensure an approximate 90° to the cutting surface. Serious degree diversion may lead to uneven thickness of the section. Next, the paraffin block is cut in a microtome to a desired thickness. The ideal section should be accomplished at a thickness of 3–5 μm, with a sharp knife blade coupled with smooth and swift cutting technique. Over thick sections, as well as sections with tears, folds, and knife nicks, may lead to false-positive or false-negative interpretations. Tissue sections should be adhered to poly-lysine pre-coated slides to avoid detachment during heat-induced epitope retrieval.
Fixation
Appropriate fixation is a vital determinant for the success of immunohistochemistry. Fixation terminates any ongoing biochemical reactions in the tissues. In addition, fixation also preserves the localisation as well as structure of the antigen to provide a target for antibodies that will be applied to detect the antigen. Neutral-buffered formalin is the most commonly and practically used fixative for routine tissue sections. Other fixatives include paraformaldehyde, Bouin’s solution, or HEPES glutamic acid buffer-mediated organic solvent protection effect (HOPE) fixative. Formalin fixes tissues by covalently cross-linking protein amino acid residues by methylene bridges. Briefly, formalin reacts with a peptide sequence to form a methylol (hydroxymethyl) group on an amino acidic residue like arginine, cysteine, lysine, tryptophan, tyrosine, serine, asparagine, or glutamine. Imine groups (also known as Schiff’s bases) are then formed by condensation of methylol groups on lysine residues. Due to its instability, imine groups condense with other components such as phenolic, cysteine, or tyrosine to form methylene bridges [143]. These cross-linkages however induce conformational alteration to the protein secondary, tertiary, and quaternary structures and block the accessibility of targeted antigenic sites to the primary antibodies. Therefore, some form of antigenicity restoration is required to increase the sensitivity of immunohistochemical markers in fixed tissues. We further discuss antigen retrieval techniques in the next section.
Optimal fixation time depends on the size of the specimen. Generally, fixation for between 18 and 24 h is recommendable for most application. Under-fixation or delayed fixation causes a gradation of immunohistochemical staining such that there is strong colourisation on the periphery yet weak signal in the core of the tissue section. This might be explained by the adequate fixation near the outside of the specimen, but that the inside of the specimen is preserved only by coagulative fixation from ethanol which would occur during tissue processing [144]. Under-fixation leads to a decrease in reliability of the immunohistochemical staining [145]. On the other side, over-fixation exaggerates the intra- and intermolecular linking that can cause poor epitope presentation, subsequently reducing immunoreactivity between antibodies and antigens [146]. Although over-fixation can mask antigenic sites, antigen retrieval can help to overcome this loss of antigen staining. In contrast, it is irretrievable to rectify under-fixed tissue and may lead to unreliable immunohistochemical results. Practically speaking, it is therefore recommended that even small specimens should be fixed for at least 6–12 h [147].
Antigen Retrieval
Most vascular specimens are fixed in formalin and subsequently embedded in paraffin. While fixation preserves tissue morphology excellently, it also modifies the structure of tissue proteins, therefore masking the targeted antigenic sites. The masking effect by fixation poses a serious obstacle for immunohistochemical detection of the antigen. There were attempts to find the alternative fixatives to replace formalin decades ago, but it seems that an ideal fixative will never be found [148]. Fortunately, the loss of antigenicity can be corrected by various methods in antigen retrieval. The general principle of antigen retrieval lies upon the breaking of protein cross-links which are induced by fixation process, with the subsequent exposition of antigenic sites in order to allow the primary antibodies to bind. Antigen retrieval is advantageous in enhancing the sensitivity of the immunohistochemical detection of epitopes. The increased immunohistochemical sensitivity allows the use of higher antibody dilutions, which lowers not only the cost but the undesirable non-specific background staining on the tissue section. It therefore minimises the possibility of false-negative results and increases the reproducibility as well as the accuracy of the interpretations. Restoration of antigens can be achieved either by physical (such as enzymatically proteolytic digestion, denaturation, or oxidising treatment) or chemical approaches (including the widely employed heat-induced epitope retrieval, or HIER). Although both approaches work by cleaving the cross-links, the choice of method depends on the antigen of interest and/or antibody to be used. It is the responsibility of the individual laboratory to compare and determine the optimal retrieval protocol for individual antigen in tissues before the staining process is carried out in bulk.
Proteolytic treatment of paraffin-embedded section was first introduced by Huang et al. [149] to detect hepatitis B core and surface antigens. Enzymatic antigen retrieval employs proteolytic enzymes to break the methylene bridges. Commonly used proteolytic enzymes include trypsin, pepsin, and pronase, but papain, bromelin, proteinase K, and collagenase are less frequently used. Due to the use of proteolytic enzymes, this retrieval method can sometimes damage the morphology as well as the targeted antigen of the tissue section. So, the kind and concentration of enzyme, and the treatment time and temperature, need to be optimised. Although enzymatic retrieval does not usually recover immunoreactions dramatically compared to heat treatment, but it is still a useful way for antigens that lose their antigenicity when exposed to heat [150].
Nowadays, heat-induced antigen retrieval is the most commonly used method. Introduced in response to the need for a more effective method to unmask the epitopes in the 1990s, this “revolutionarily new” technique yielded satisfactory outcomes and it was widely employed in pathology [151]. Heat-induced antigen retrieval can be performed using a pressure cooker, a scientific microwave, a vegetable steamer, an autoclave, or in water bath. Volkin and Klibanov [152] suggested that heat would reverse the protein conformation altered by fixation, inducing a “renaturation” of the epitopes. Since the mechanism of heat-induced antigen retrieval relies on the cleavage of the cross-links and in the subsequent structural reconstruction of the antigen, all the factors involved in the procedure, which can potentially influence the antigenicity restoration, need to be controlled to guarantee the retrieval efficacy. The factors include pH, molarity, and type of the retrieval buffer, dilution of primary antibody, and treatment time. The most important factor in retrieval efficiency is the heat. Most protocols suggest antigen retrieval time for 10–20 min at about boiling temperature. However, it is only a suggested time. Under-retrieval may lead to weak staining, but over-retrieval may cause excessive non-specific background staining as well as increase the occurrence of tissue detachment from the slides, especially when it is a minute aortic section.
The next influential factor in antigen retrieval would be the choice of retrieval buffer. Generally, sodium citrate buffer at acidic pH 6.0 and Tris-EDTA buffer at basic pH 9.0 are the most popularly used retrieval solutions for the majority of antigens. Although there is no universally optimal pH for antigen retrieval and the pH choice depends on the targeted antigen, a study has elegantly demonstrated the important role of retrieval solution pH in the process of retrieving antigen [153]. The generation of electrostatic repulsion and hydrophobic attractive forces between antigen molecules in acidic or basic medium might explain the improved immunoreaction. Antigen extends and exposes its hydrophobic and hydrophilic portions after the destruction of methylene bridges by heat (Emoto et al. 2005). This is the very moment where pH fundamentally influences the outcome of the retrieval process. At neutral pH, the antigens are entangled together due to the simultaneous presence of hydrophobic and ionic forces. At acidic or basic pH however, the antigens are charged negatively or positively to create two opposing forces that cooperatively prevent entangling of the protein and thus “open up” the antigenic sites. Kim et al. [153] found that increasing buffer pH from 6.0 to 9.0 yielded gradually improved retrieved antigen immunoreactivity against most antibodies. This may be due to the fact that acidic hydrolysis is much more complex and has a more rate-limiting step than basic hydrolysis. Therefore, in order to avoid under- or over-retrieval, we recommend a control experiment beforehand to evaluate the optimal retrieval protocol for a particular primary antibody, where a known positive tissue control for that antibody is retrieved in either sodium citrate or Tris-EDTA buffer for a range of time at 5 min intervals. At the same time, different dilutions of the antibody are tested. An optimised antibody dilution should produce the highest colour intensity of positive staining with the non-specific background staining kept at the lowest. It is always advisable to start by using the antibody at the dilution recommended on the supplier’s technical datasheet.
Briefly, the optimisation method for mouse monoclonal anti-rat intercellular adhesion molecule-1 (ICAM-1) antibody as an example is discussed here. Human tonsil is the positive tissue control for ICAM-1 as recommended on the product datasheet. Even after being microwave heated for more than 10 min in sodium citrate buffer at pH 6.0, the brownish staining in lymphatic nodules, where ICAM-1 is abundantly expressed, was still absent or weak (Fig. 8.4a–c). This might be due to the low concentration of antibody (1:100) that had been applied. So, the dilution of antibody was decreased to 1:50. However, the staining was still weak regardless of how long the sections were heated (Fig. 8.4d–f). At this point, the results suggested that pH 6.0 might not be suitable for the antibody and/or antigen. Then, the sections were retrieved in Tris-EDTA pH 9.0 buffer for various durations. Results showed that retrieval for 20 min caused the tonsil section to be over-retrieved, leading to colour over-saturation and undesirable background staining around the lymphatic nodules (Fig. 8.4i). Surprisingly, retrieval for 15 min constantly gave a rather weaker staining compared to 10 min (Fig. 8.4h). Retrieval for 10 min produced satisfactory staining (Fig. 8.4g). The results were in agreement with a previous study by Kim et al. [153] reporting that basic buffers yielded more satisfactory immunostaining than the acidic buffers. These findings also emphasise the importance of optimising the pH of retrieval solution before immunostaining the samples. Table 8.1 shows the suggested optimal conditions for different antibodies. Please note that they are only suggested methods. Individual laboratory needs to find out the optimal retrieval protocol for individual antigen in certain tissues before the staining process is carried out in large amount.
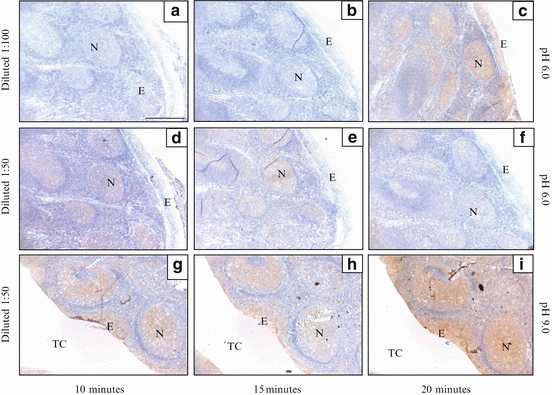
Fig. 8.4
Photomicrographs showing immunohistochemical optimisation for mouse monoclonal antibody anti-rat ICAM-1 on human tonsil sections. The sections are incubated with antibody at dilution factor of 1:100 or 1:50 for an hour at room temperature after being heat-retrieved in either sodium citrate pH 6.0 buffer or Tris-EDTA pH 9.0 buffer, for 10, 15 or 20 minutes. Brownish colour in lymphatic nodules (N) indicates positive staining for ICAM-1 antigens. The optimal condition is chosen based on the colourisation. The sample retrieved in pH 6.0 buffer (a & b) does not react well with the antibody (diluted 1:100) until it is heated for 20 minutes (c). When the concentration of the antibody is increased, it produces satisfactory colour after only a 15 minutes retrieval (d). However, the reaction becomes poorer when the sample is heated longer than that (e & f). When the pH of the retrieval buffer increases to pH 9.0, it gives good staining after 10 or 15 minutes heating (g & h). But heating for 20 minutes yields oversaturation colour (i). Abbreviations: E, epithelium; TC, tonsillar crypt. (Magnification: 4×; scale bar: 500 μm)
Table 8.1
Suggestions of optimal conditions for immunohistochemical staining of inflammatory biomarkers
Antibody (anti-) | Fixative | Retrieval buffer | Antibody dilution | Heat-induced retrieval time | Tissue | References |
|---|---|---|---|---|---|---|
VCAM-1 | 10 % neutral-buffered formalin | Sodium citrate pH 6.0 | 1:100 | 10 min | Aorta | Ng et al. [23] |
ICAM-1 | 10 % neutral-buffered formalin | Tris-EDTA pH 9.0 | 1:50 | 10 min | Aorta | Ng et al. [23] |
LOX-1 | 10 % neutral-buffered formalin | Sodium citrate pH 6.0 | 1:100 | 10 min | Aorta | Ng et al. [23] |
P-selectin | 4 % paraformaldehyde | 0.037 % EDTA/ 0.055 % Tris buffer | 1:50 | 5 min | Artery | Dever et al. [68] |
E-selectin | 10 % formalin | EDTA buffer pH 8.0 | 1:100 | 10 min | Testes | Sukhotnik et al. [154] |
ETA receptor | 10 % buffered formalin | Citrate buffer pH 6.0 | 1:100 | 10 min | Pulmonary artery | Ishida et al. [155] |
ETB receptor | Paraformaldehyde | Citric acid buffer pH 6.0 | 1:200 | 20 min | Mesenteric artery | Zhang et al. [96] |
TNF-α | Buffered formalin | Citrate buffer pH 6.0 | 1:200 | 18 min | Plantar section | Nobre et al. [156] |
VEGF | 10 % phosphate-buffered paraformaldehyde | Citrate buffer pH 6.0 | – | 15 min | Heart | Zaitone and Abo-Gresha [157] |
iNOS | 10 % phosphate-buffered paraformaldehyde | Citrate buffer pH 6.0 | – | 15 min | Heart | Zaitone and Abo-Gresha [157] |
Ang II | Formalin | Citrate buffer pH 6.0 | 1:500 | 5 min (for three times) | Ovary | Harata et al. [158] |
AT1R | 4 % paraformaldehyde | Citrate buffer pH 6.0 | 1:150 | – | Brain | Thomas and Lemmer [159] |
Immunohistochemical Staining
There are numerous immunohistochemical staining techniques that vary from simple one-step procedures, where the label is directly linked to the primary antibody, i.e. direct methods, to complex multiple-step procedures, where the label is conjugated to a secondary antibody or a new micropolymer labelling technology, i.e. indirect methods. Although indirect methods are more complicated and consume more time than direct methods, the indirect methods are recommended on vascular tissues because the methods generally yield very sensitive and reproducible results. Other researchers also prefer indirect methods over direct-labelled antibody methods [160,161]. This is especially useful when the protein of interest is expressed low in the specimen. Of the labels conjugated to a secondary detector, horseradish peroxidase (HRP) and alkaline phosphatase are the most commonly used enzymes. A number of chromogens or substrates for these enzymes including 3,3′-diaminobenzidine (DAB), 3-amino-9-ethyl carbazol (AEC), and 5-bromo-4-chloro-3-indoyl phosphate/nitroblue tetrazolium (BCIP/NBT) are used to visualise the antigen–antibody binding. Chromogens DAB and AEC produce intense and contrasting colourisation on the tissues. However, the pitfall in these staining systems would be the endogenous peroxidase activity that can lead to false-positive staining. To avoid this, one should first use hydrogen peroxide to suppress the endogenous peroxidase activity in the tissue. A blocking step also helps to reduce the background staining [161].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


