Fig. 5.1
Identification of target proteins using IHC on tissue sections or cytological preparation slides. HRP/DAB detection method was used for all images except C. (a) Endothelin A expression on human prostate cancer tissue. (b) Phospho-c-Myc expression in the nuclei of human kidney carcinoma tissue. Insert: enlarged image. (c) Multi-fluorophore detection of target proteins on HCT-15 human colon cancer cells, spiked into healthy subject PBMC. Tumor cell heterogeneity is visible. Green—Alexa 488 conjugated anti-cytokeratin 8/18 (CK8/18) monoclonal Ab; red—Alexa 594 conjugated anti-epithelial platelet cell adhesion molecule (EpCAM) monoclonal Ab; orange—Alexa 555 conjugated anti-breast cancer 1 (BRCA1) polyclonal antibody; blue—DAPI. (d) Human cancer tissue microarray stained with polyclonal anti-Bcl-XL antibody. HCC hepatocellular carcinoma, CRC colon rectal carcinoma, PC prostate cancer, HL Hodgkin’s lymphoma
Basic Procedure of IHC
IHC is typically performed on 4–6 μm thick formalin-fixed paraffin-embedded (FFPE) tissue slices or a justified thickness (8–90 μm) of frozen fresh tissue. Through a series of incubation steps the primary antibody specifically binds the target protein and is visualized using an enzyme-linked chromagen or fluorophore conjugate.
Fixation
To adequately preserve cellular proteins and avoid tissue autolysis after sample collection, tissues need to be fixed [3, 23]. Two types of fixatives are used in histopathology: cross-linking (non-coagulating) fixatives and coagulating fixatives. A commonly used cross-link fixative is formaldehyde, which preserves mainly peptides and the general structure of cellular organelles for histopathological assessment and IHC interpretation. The mechanism of fixation with formaldehyde is the formation of cross-links between the formalin and uncharged reactive amino groups (–NH or NH2). The cross-links result in a profound change in the conformation of macromolecules and allow recognition of proteins (Ag) by antibodies [24]. Formaldehyde fixation is a progressive time- and temperature-dependent process. Overfixation commonly occurs during the FFPE process and can produce false-negative results from excessive cross-links [23]. Therefore, an Ag retrieval procedure to restore the protein (Ag) configuration is commonly used to avoid false-negative results for FFPE tissue sections. In our tumor vascular study, the tumor samples were processed by either the FFPE process or the frozen fresh tissue process. For FFPE, the 5 mm3 tumor samples were collected and immediately immersed in 10 % neutral formalin for at least 2 h prior to the FFPE process. For the frozen procedure, 5 mm3 to 1 cm3 fresh tumor samples were collected and immediately snap frozen by immersing in liquid nitrogen. Samples were kept for up to 2 h in liquid nitrogen as samples were collected, and then all samples were transferred to an −80 °C freezer. Cryosections were generated from a −20 °C cryostat and fixed in 4 % paraformaldehyde, pH 7.4, for 1 h, washed in PBS, air dried, and stored at 4 °C.
For the circulating tumor cell study, a light formaldehyde fixation (1 %, for 5 min) was used for cytological slide preparation from fresh samples, followed by a PBS buffer rinse and air dry. Slides were stored at −80 °C with desiccants for up to 1 year.
FFPE Tissue Section and Frozen Fresh Tissue Cryosection
IHC assays are performed on tissues in various forms, including FFPE tissues, frozen fresh tissues, and cytological preparations or smears. The choice between the various preparations depends on research needs for preservation of cell/tissue morphology and duration of storage. FFPE tissue blocks provide optimal morphology and are easy to handle and store. For general morphological assessment and IHC protein evaluation, 4–6 μm FFPE sections are commonly used. However, the antigenicity may decrease with longer storage times; the Ag retrieval procedure can help to restore the antigenicity. Cryosections provide better antigenicity of proteins with rapid turnover times but require quick procedures such as snap frozen tissues, use of a low temperature cryostat for sectioning, and immediate fixation. If a frozen tissue is not sectioned carefully or the tissue blocks are stored for long durations, poor tissue morphology may result. In our vascular study, for the purpose of generating data rapidly to accommodate drug discovery timelines, we used the frozen fresh tumor process for most of the tumor vascular studies. 90 μm thickness cryosections were obtained for global tumor vascular observation and 20 μm thickness cryosections, recommended for better length of microvessel observation, were obtained for the identification of target proteins such as the phosphorylated receptors for PDGF and VEGF, microvessels, pericytes, and hypoxic proteins. We particularly chose 20 μm thicknesses for the study because the vasculature presented good morphology not only in the length of the vessel but also pericyte coverage, which provided additional information for the development of linifanib [25, 26].
Tissue Pretreatment and Antigen Retrieval
The effects of formaldehyde can be partially reversed [27] to restore antigenic epitopes of the target protein prior to antibody binding [1, 28]. In general, several different tissue pretreatments for FFPE tissue sections should be tested to determine which approach provides the best access of the antibody to the antigen epitopes. Pretreatment options include heat-induced epitope retrieval (HIER ) [29] and enzymatic treatment with proteolytic enzymes, which presumably breaks the formalin-induced methylene cross-links in the antigenic molecules to restore their immunoreactivity. In the cancer biomarker studies, to unmask antigenicity of pPDGFR β, pVEGFR 2, von Willebrand factor (vWF), C-myc, endothelin A (ETA), and Bcl-xl proteins from FFPE tissue sections, the pretreatment procedure was performed after tissue sections were dewaxed and rehydrated by a graded alcohols procedure. The tissue sections were heated in the presence of Tris buffer pH 9 solutions as a Target Retrieval Solution using a pressure cooker at 120 °C for 2 min (Digital Programmable Pressure Cooker, Biocare Medical, Walnut Creek, CA). In the absence of pretreatment, the target proteins could not be detected or were poorly detected by the respective antibody, demonstrating the need in this case of using an antigen retrieval procedure (Fig. 5.2). There would be no need to use this procedure with frozen fresh tissue cryosections.
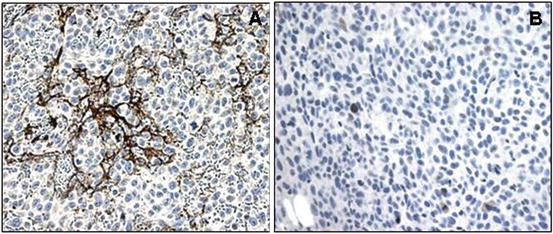
Fig. 5.2
vWF antibody immunostaining on the HT1080 human xenograft fibrosarcoma tissue to detect endothelial cells. (a) IHC staining without antigen retrieval. No vessels are visible. (b) IHC staining after antigen retrieval (HIER). Characteristic tumor vasculature including dilated, saccular, and disorganized vessels is notable. HRP/DAB detection method. Hematoxylin counterstain
Blocking Considerations
Protein blocking
The blocking solution contains nonspecific proteins isolated from the species in which the antibody was made to prevent nonspecific binding of the primary or secondary antibodies. The blocking reagent can also comprise serum-free recombinant blocking proteins, and is used universally for any detection system. The blocking step usually includes an incubation period for approximately 5–10 min prior to the antibody incubation. We used a serum-free blocking reagent for 5 min before the primary Ab incubation and the results were satisfactory (Figs. 5.1 and 5.2).
Enzyme blocking
Endogenous peroxidase in blood cells can react with DAB (3′, 3′-diaminobenzidine) to produce a brown product indistinguishable from specific immunostaining using the HRP/DAB (horseradish peroxidase/DAB) detection system [1]. The native peroxidase activity in tissue sections can be destroyed almost completely using a diluted solution of hydrogen peroxide (0.03–3 % H2O2). In our tumor vascular IHC studies, 3 % hydrogen peroxide was used to ensure the xenograft tumor tissues, which had abundant hemorrhages, had clean backgrounds. The tissue sections were incubated in 3 % hydrogen peroxide for 5 min before the primary antibody incubation, and buffer rinses were performed after primary antibody incubation. The buffer washes must be performed before the HRP incubation.
There is another commonly used chromagenic detection system, alkaline phosphatase/fast red, in which 2–3 % levamisol is used to block endogenous alkaline phosphatase.
Fc blocking
Fc receptors of mononuclear blood cells are usually destroyed during FFPE procedure but can cause nonspecific binding of the primary antibody to lightly fixed frozen tissues [1, 3]. During the tumor vascular study, we used 4 % paraformaldehyde to fix 20 μm frozen sections for an hour and stained with individual antibodies that were directly conjugated with fluorophores, including pPDGFR β, CD31, α-SMA, and collagen IV. The background was at an acceptable level without Fc blocking step (Fig. 5.3).
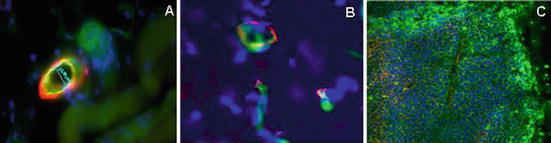
Fig. 5.3
Multi-fluorophore labeling used in pericyte coverage and microvessel measurements. 20 μm cryosections stained with various antibodies in the absence of an Fc blocking step. (a) Dual labeled microvessel and pericyte on HT1080 human fibrosarcoma tumor-bearing mouse skeletal muscle. Lectin/FITC perfusion for detection of vessels (green) and polyclonal Ab α-SMA/Alexa 594 staining for pericytes (red). (b) Dual labeling of vessel and basement membrane on normal rat brain tissue. Lectin/FITC perfusion for detection of vessels (green) and polyclonal Ab collagen IV/Alexa 594 staining for structural protein expression in matrix around vessels. (c) Multi-fluorophore labeling of SW620 human colon tumor tissue. Drug distribution assessed by i.p. administration of Paclitaxel/Oregon green to tumor-bearing mouse. Hypoxic protein expression was detected by mIgG/Alexa 647 conjugate (yellow). Lectin/Alexa 594 perfusion for microvessels. Blue—nuclei/DAPI
Primary Antibody and Controls
In IHC, the primary Ab is used to bind and recognize a specific antigen in tissue sections or cytology slides. Primary Abs can be monoclonal or polyclonal and made in diverse species. One requirement for optimization of an IHC assay is to establish that the antibody binds only to the protein containing the immunogen peptide. Although each Ab is generated against a particular immunogen, dilution of the primary antibody still needs to be optimized with appropriately chosen controls. Controls for the specificity of an antibody are important for the interpretation of protein localization and expression [30]. Specific controls for assay development comprise positive and negative controls. The positive controls use cells or tissues known to contain the protein. The negative controls use cells or tissues known not to contain the protein, or replacing the primary antibody with serum or an isotype control antibody and lastly a preabsorption control, in which the antibody is mixed with the protein or peptide used to generate the antibody. Primary antibody incubation times and temperatures range from an hour at room temperature or 37 °C to overnight at 4 °C. The working concentration of primary Ab for IHC assay generally ranges from 2 to 10 μg/mL. After incubation is completed, the tissue sections should be rinsed thoroughly to remove excess Ab.
In linifanib development, the Abs we used for vascular biomarker identification, including pPDGFR β, pVEGFR 2, vWF, CD31, α-smooth muscle actin (α-SMA), collagen IV, and hypoxic protein, are commercially available. Each antibody was generated against synthetic peptides and the specificities determined by immunoblot and/or immunoprecipitation, greatly reducing the possibility of binding to epitopes not found on the original peptides [25, 26, 31]. The primary antibodies were incubated with tissues for 2 h at room temperature with gentle horizontal rotation or stored at 4 °C overnight. Matched isotype control antibodies for all primary antibodies and preabsorption controls for pPDGFR β and pVEGFR 2 were used.
Detection Systems
Detection systems are classified as direct and indirect. Each method uses a label conjugated to the primary Ab, secondary Ab, or tertiary Ab to allow visualization of the tissue Ag–Ab immune complexes with microscopy. The detection system needs to be carefully selected during assay development taking into consideration the Ab specificity, color discrimination, cell type, and tissue origin. A variety of labels have been used, including fluorescent compounds, enzymes, and metals [32].
Direct Methods
Direct methods of detection are straightforward, based on the conjugation of the primary antibody to a label. Different labels that have been used include fluorochromes, enzymes, colloidal gold, and biotin [3]. With direct methods, the prepared tissue sample is incubated with antibody-label conjugate. Examples of fluochrome labeling are the Alexa dyes for fluorescence detection. One indirect method but referred to as a specific method is the Zenon labeling system (Invitrogen, Grand Island, NY). Zenon labeling technology utilizes a fluorophore-, biotin-, or enzyme-labeled Fab fragment directed against the Fc domain of the primary antibody. Fab fragments specific for human or mouse IgG1, IgG2a, IGg2b, and rabbit IgG are available. The labeling method does not require purification of the labeled antibody and intra-species cross reactivity is low. Since the Fab fragment is provided as a fluorophore conjugate, we were able to have quick turnaround times for assay development, avoid nonspecific antibody binding, and obtain sufficient signals for imaging data analysis for the tissue-based tumor vascular study and the cell-based CTC study. Moreover, this technology permits the use of multiple antibodies in a single staining procedure. Each antibody is labeled with a different.
Indirect Method
An indirect system increases the sensitivity of detection by introducing a labeled secondary antibody which recognizes the primary antibody. If the primary Ab is made in rabbit, the secondary should bind to the rabbit Ab. The secondary antibody is labeled with an enzyme, such as HRP or fluorescent dye, includes a polymer that increases the sensitivity of the detection system, and reduces the background staining [33]. Multiple molecules of enzyme and the secondary antibody are attached to this polymer, which simplifies the three-step methods such as ABC (avidin–biotin–peroxidase complex) while maintaining equal or higher sensitivity.
The sensitivity of this method is higher than a direct method due to signal amplification: the increased ratio of label to primary antibody increases the intensity of the reaction [3]. However, specificity may be lower because of nonspecific binding by the secondary Ab. We used a commercially available polymer-based labeling two-step method in the HRP/DAB chromagenic detection system to detect diverse target proteins (Fig. 5.1). The incubation time was 20–30 min and PBS buffer was used to rinse prior to the use of a chromagen.
Combination of Detection Systems for Multiple Target Protein Detection
Different IHC detection methods can be incorporated into a single protocol to simultaneously visualize multiple components. For example, antibody-based detection of antigen epitopes can be combined with non-antibody-based methods such as lectin binding to glycoproteins on surface of endothelial cell. One such protocol is detailed in section “Assay Development”.
Chromagens
DAB is often used as chromagen for HRP-labeled antibodies. DAB reacts with the peroxidase enzyme in the presence of hydrogen peroxide and is converted to a dark brown insoluble precipitate which provides high resolution. The incubation time in DAB is 3–5 min, while the negative control is checked to obtain the necessary high quality of resolution. Buffer rinses are performed after incubation is complete. Fast red is a commonly used dye for the alkaline phosphatase (ALP) detection system. Fluorescent labeled secondary antibodies can also be used to boost signals.
Counterstaining
Counterstaining is the final step in IHC. It enhances the morphology and contrast of cells and tissues. Hematoxylin is the most commonly used counterstain in chromagenic detection methods. The molecule binds to the acidic components in nuclei, resulting in a purplish-blue stain. The duration of exposure to hematoxylin is approximately 30 s to 1 min, based on the color appearance. 0.8 % ammonium chloride or PBS buffer rinse can speed the nuclei blue process. The sample is rinsed with water after the process is complete. Slides can be air dried prior to placement of a coverslip.
Fluorescence stained slides are washed with PBS, rinsed with distilled H2O, air dried, and covered with prolong anti-fade mounting medium containing DAPI (4′, 6-diamidino-2-phenyindole, Invitrogen), which immediately stains the nuclear DNA fragments.
Coverslip
When the IHC process is complete, the stained slides should be covered by mounting medium for microscopic examination and storage. Several applications are commercially available to meet the purpose. Usually, chromagen stained slides are dehydrated through a series of increasing alcohol concentrations, rinsed in xylenes, and coverslipped in permanent mounting medium for long storage. In our studies, we used aqueous mounting medium (Sigma, St. Louis, MO) to mount both tissue slides and cell slides directly after air drying. Similar results were obtained without dehydration using alcohol. For fluorophore detection, a mounting medium containing DAPI as described above was used to coverslip after slides were air dried. The results were satisfactory.
IHC for Cancer Biomarker Studies
Cancer biomarker studies using IHC for target protein identification can answer three questions: can we identify useful targets by differential cell expression, is our target present on the cells of interest, and is our protein of interest modified by treatment. Our tumor vascular study and circulating tumor cell study elaborated the utilization of IHC in oncology drug development.
Cancer Biomarker Studies on Tissue Section Slides
IHC localizes a target protein at the microscopic level, which allows us to better understand the cellular expression pattern in normal and disease tissue as well as the subcellular localization pattern. In oncology drug early development programs, tests are generally performed in xenograft tumor models to evaluate whether particular target proteins may be present as biomarkers in clinical samples. The preclinical studies provide baseline information for the therapeutic effect in tumor cells. With IHC, the cell-specific expression pattern of the marker within the tumor can be determined; results from such studies have been used to make go/no go decisions [34]. For oncology drug development at Abbott (AbbVie), IHC assays provide cell-specific detection of the target protein and information on what cell type expresses the target. The specific target protein expression, or change of expression level and localization, has helped us to understand the biological response to oncology drug candidates (Figs. 5.1 and 5.4). Particularly in linifanib development, the IHC studies on target proteins phospho-VEGFR 2, which is expressed predominantly in endothelial cells and tumor cells, and phospho-PDGFR β, which is expressed primarily in pericytes and tumor cells, in xenograft tumor models of HT1080 (human fibrosarcoma), SW620 (human colon carcinoma), and 9L (rat glioma), demonstrated that treatment with linifanib strongly inhibited the immunostaining with both phosphorylation-specific antibodies to the receptors. From this data, we concluded that inhibition of the primary angiogenic targets in vivo (Fig. 5.4a) correlated with tumor growth inhibition [25, 26].
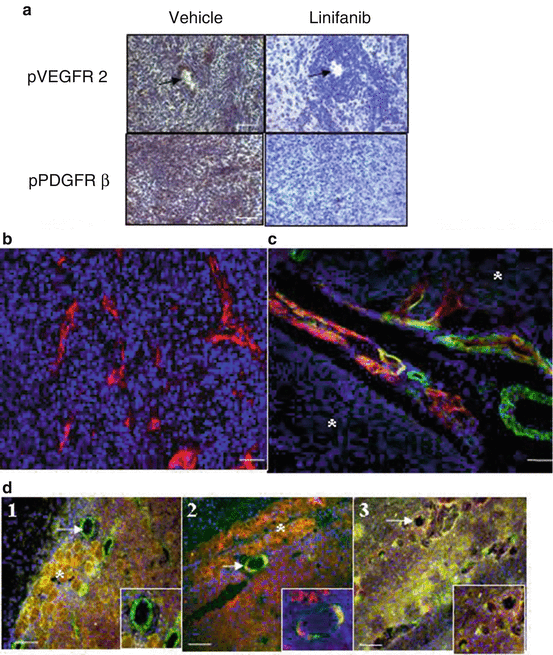
Fig. 5.4
Tumor vascular identification in antiangiogenic drug development. (a) pPDGFR β and pVEGFR 2 immunostaining on 9L rat xenograft glioma tissue using HRP/DAB detection method. Linifanib inhibited phosphorylation of both receptors on endothelial cells and tumor cells. Arrows indicate microvessels. (b) U87-MG human xenograft glioma tumor tissue stained with CD31/Alexa 594 stained for microvessels (red). (c) Dual fluorophore labeling of vessels and pericytes in HT1080 fibrosarcoma tissue. Endothelial cells (red) form vessels from the host tissue into the tumor; pericyte (green) coverage follows. Lectin/Alexa 594 vascular perfusion for vessels (red) and α-SMA/Alexa 488 staining for pericyte coverage (green). Asterisks: Tumor islet. (d) Dual labeling of α-SMA/FITC (green) and pPDGFR β/Alexa 594 (red) on HT1080 fibrosarcoma tissue demonstrated co-localization of the two proteins. (d1) No co-localization was detectable on a vessel located in host muscle tissue. (d2) Some co-localization is visible on a vessel located between host and tumor tissue. (d3) Co-localization presented on vessels in tumor area. Asterisks: Host tissue. Arrows point vessels. Blue indicates nuclei for all images. Bars represent 50 μm in a, b, and d, 20 μm in c
Cancer Biomarker Study on Cytological Preparation Slides
Immunocytochemistry (ICC) is frequently used to characterize circulating tumor cells (CTC) as potential biomarkers. After the CTC population is enriched and placed on slide, data derived from enumeration and characterization of CTC using ICC have the potential to stratify cancer patients for outcome and to provide information on patient response to therapy [35–37] Sensitive and reproducible ICC assays are required to identify the patients most likely to benefit from the treatment. Accurate results from ICC are challenging to obtain, particularly for CTC due to their low abundance in blood and because multiple cell surface markers are ordinarily required to correctly identify a CTC. Our CTC study combined the classification of immunostaining pattern in diverse blood nucleated cells with cell morphological assessment. To detect and enumerate CTC, multi-fluorophore labeling is required to exclude non-CTC cell populations, mostly leukocytes, and identify tumor marker(s) expressed on CTC (Fig. 5.1c). Since CTC are rare in blood, ranging from zero to thousands per mL of blood, an automated scanning system for immunostained slides and software for image analysis are also required [38, 39].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


