DIAGNOSIS
Congenital heart disease encompasses a wide range of anomalies that result from abnormal fetal development of the heart. Defects can range from simple to complex. The age of presentation of these defects depends primarily on the physiologic impact of the anomaly. After birth, patients can present within minutes to hours with profound hypoxemia or hemodynamic collapse. Others may present weeks to months later with evidence of a new murmur or signs of congestive heart failure. Relatively asymptomatic lesions can go undetected until children are school age or adolescents. With current ultrasound imaging, many cardiac anomalies are identified on prenatal examination.
Early and accurate diagnosis of congenital heart disease requires careful identification of signs and symptoms of heart disease. The initial workup begins with a focused history and physical examination. Classification of heart murmurs can be highly suggestive of underlying cardiac anomalies. Early signs of heart disease include cyanosis, tachypnea, unequal pulses, and failure to thrive. Key symptoms of congenital heart disease in the patient’s history include feeding difficulties, irritability, and frequent respiratory infections.
Standard diagnostic studies include a chest radiograph (CXR) and electrocardiography (EKG). Some cardiac defects have pathognomonic findings on CXR. Other anomalies may be suspected based on heart size, increased or decreased pulmonary markings, aortic arch sidedness, or situs abnormalities (the heart located in the mid- or right chest rather than the typical location in the left chest). EKG can identify rhythm disturbances, axis deviation, atrial enlargement, and ventricular hypertrophy. Transthoracic echocardiography is often the only diagnostic test needed to provide the anatomic detail required for surgical planning. Cardiac catheterization, cardiac magnetic resonance imaging (cMRI), and computed tomography angiography (CTA) are used as adjunct diagnostic tests when additional information on flow, pressure, resistance, or anatomic detail is required.
PREOPERATIVE MANAGEMENT
The care of congenital heart disease patients requires a collaborative effort of a multidisciplinary team of cardiologists, surgeons, interventionalists, echocardiographers, and radiologists. For the majority of congenital heart defects, surgical correction or catheter-based intervention is necessary for definitive treatment. Careful timing and planning of operative and catheter-based interventions along with highly skilled preoperative and postoperative care are essential to a successful outcome.
Certain defects such as atrial septal defects (ASDs), ventricular septal defects (VSDs), and patent ductus arteriosus (PDA) may resolve spontaneously over the first several years of life. The remaining defects require intervention when the risk of surgery is reasonable, when symptoms can no longer be managed medically, and/or prior to the onset of irreversible complications.
Neonates presenting with ductal-dependent lesions require ductal blood flow to maintain systemic or pulmonary perfusion. Ductal patency is achieved with intravenous prostaglandin E1 therapy. Supplemental oxygen is supplied only as necessary for cyanosis as newborns tolerate relative cyanosis (oxygen saturations > 70%) quite well. The remainder of therapy is directed toward managing congestive heart failure symptoms with diuretics, afterload reduction, and maximal caloric intake.
OPERATIVE MANAGEMENT
For most congenital heart defects, surgical correction is possible. For more complex defects, staged repair with early palliation as well as staged palliation remain options. The anticipated somatic growth of the child must be considered when determining the surgical approach. Invasive monitoring lines are essential for monitoring patients during surgery and postoperatively. All patients have arterial and central venous catheters placed along with peripheral intravenous catheters and a Foley catheter. For neonates, the umbilical vessels are the desired venous and arterial access. Avoidance of long-standing and repeated femoral lines is important because patency of these vessels is often essential for diagnostic and interventional catheter procedures later in life. Due to variable blood flow with perfusion techniques and underlying cardiac anomalies, it is possible to have differential cooling and rewarming. Therefore, temperature probes are placed in the nasopharynx, the rectum, and on the skin to allow for accurate monitoring.
Most surgical repairs require cardiopulmonary bypass. This involves drainage of venous blood from the patient via cannulae placed in the superior and inferior vena cava (for intracardiac repairs) or a single cannula in the right atrium. Blood passes through the bypass circuit, which warms or cools the blood to the desired temperature, adds oxygen and removes carbon dioxide, and pumps the blood back into the body via an arterial cannula, usually in the ascending aorta. Hypothermia may be employed to decrease the metabolic demands of the body and heart, which provides additional protection against ischemia. The degree of hypothermia, 18-34°C, depends on the complexity and time needed to complete the procedure. The heart can be arrested with high-potassium cardioplegic solution delivered through the coronary arteries antegrade (through a cannula proximal to an aortic cross-clamp in the aorta) or retrograde (through a cannula placed in the coronary sinus). Arresting the heart allows the surgeon to operate safely with a still and bloodless field. An additional vent can be placed via the right superior pulmonary vein to capture pulmonary venous return and to assist in deairing of the heart.
Hypothermic circulatory arrest is required for complex aortic arch reconstructions. This involves cooling the patient to 18°C for a minimum of 20 minutes to ensure even cooling of the brain and body. The head is packed in ice, the patient’s blood is drained into the venous reservoir, and the pump is turned off. The cannulae can then be removed from the field to aid in visualization and repair of the arch. No absolute safe duration of hypothermic circulatory arrest has been determined, but it is generally felt that it should be limited to no more than 45 minutes. Alternative techniques, such as regional cerebral perfusion and intermittent low-flow perfusion, have been employed to minimize the need for hypothermic circulatory arrest. To date, however, there is no literature to support that these techniques have additional benefit without additional risk.
POSTOPERATIVE MANAGEMENT
Patients are brought to the intensive care unit intubated and mechanically ventilated. All patients have temporary pacing wires in place for management of bradyarrhythmias and tachyarrhythmias. Many patients have additional intracardiac lines in place for pulmonary artery and left atrial pressure monitoring. Drainage catheters are placed within the mediastinum to prevent accumulation of blood and fluid. These are usually removed 2-4 days following surgery. Prophylactic antibiotics are given preoperatively as well as postoperatively when drainage tubes are in place.
Cardiopulmonary bypass produces a significant inflammatory response due to activation of cytokines. Patients exhibit fluid retention and pulmonary dysfunction as a result, requiring aggressive use of diuretics and mechanical ventilation as needed. Bleeding is a common complication following cardiac surgery and infrequently requires surgical exploration (< 2%). Postoperative coagulopathy results from a multitude of factors, including hemodilution, platelet damage, factor consumption, immature hepatic production of clotting factors, and incomplete reversal of heparin with protamine sulfate. Approximately 30% of all patients will have some arrhythmia after surgery, ranging from simple premature ventricular contractions to malignant tachyarrhythmias. The risk for long-term arrhythmias requiring chronic medication or heart block requiring a permanent pacemaker is approximately 1%. Most patients require hemodynamic support with vasopressors along with afterload reduction as necessary for ventricular dysfunction. Dopamine, epinephrine, and vasopressin are the first-line vasopressors for pediatric patients. Milrinone is used primarily for afterload reduction. Critically ill neonates may have thyroid and adrenal hypofunction, which can further exacerbate postoperative hemodynamic instability. Profound hemodynamic compromise occasionally requires additional mechanical assistance. Extracorporeal membrane oxygenation (ECMO) is the most widely and acutely available mechanical support for the pediatric population. The survival for congenital heart disease patients requiring ECMO support is approximately 50%. For patients in low cardiac output state, it is essential to rule out residual defects or repair failures that could be readdressed surgically or in the catheterization laboratory.
The pediatric population has highly reactive pulmonary vasculature that is unique from the adult cardiac surgery population. Postoperative pulmonary hypertensive crises can occur in the newborn and infant population. Crises can be initiated with agitation such as endotracheal suctioning. Maneuvers to minimize and treat pulmonary hypertensive crises include high-dose opioid anesthesia with fentanyl, paralysis, respiratory alkalosis, high fraction of inspired oxygen, and inhaled nitric oxide. Chronic agents to treat persistent pulmonary hypertension include phosphodiesterase inhibitors (eg, sildenafil) and prostacyclin (eg, Flolan).
CYANOTIC HEART DEFECTS
Cyanotic heart defects result from shunting of deoxygenated blood from the right side of the heart to the oxygenated left side of the heart or inadequate pulmonary blood flow. The relative mixing of deoxygenated and oxygenated blood produces desaturation of the arterial blood. The majority of cyanotic heart defects are diagnosed within the first few days to months of life. Cyanotic heart defects represent approximately 25% of all congenital heart defects.
The classic 5 T’s of cyanotic heart defects—(1) tetralogy of Fallot (TOF); (2) transposition of the great arteries (TGAs); (3) truncus arteriosus; (4) total anomalous pulmonary venous return; and (5) tricuspid atresia are presented in this chapter along with hypoplastic left heart syndrome (HLHS).
TOF is the most common cyanotic congenital heart defect. It occurs in 0.6 per 1000 live births and has a prevalence of about 4% among all patients with congenital heart disease. The pathologic anatomy is frequently described as having four components: VSD, overriding aorta, pulmonary stenosis, and right ventricular (RV) hypertrophy (Figure 19–27). Embryologically, the anatomy of TOF is thought to result from a single defect: anterior malalignment of the infundibular septum. The infundibular septum normally separates the primitive outflow tracts and fuses with the ventricular septum. Anterior malalignment of the infundibular septum creates a VSD due to failure of fusion with the ventricular septum and also displaces the aorta over the VSD and right ventricle. Infundibular malalignment also crowds the RV outflow tract, causing pulmonary stenosis and, secondarily, RV hypertrophy. Prominent muscle bands also extend from the septal insertion of the infundibular septum to the RV free wall and contribute to the obstruction of the RV outflow tract. The pulmonary valve is usually stenotic and is bicuspid in 58% of cases. Pulmonary atresia occurs in about 7% of cases. The branch pulmonary arteries in TOF may exhibit mild diffuse hypoplasia or discrete stenosis (most frequently of the left pulmonary artery at the site of ductal insertion). Coronary artery anomalies are frequently present. The origin of the left anterior descending artery from the right coronary artery, which occurs in 5% of cases, is clinically important because the vessel crosses the RV infundibulum and is vulnerable to injury at the time of surgery. A right aortic arch is present in 25% of patients. Associated defects include ASD, complete atrioventricular septal defect (AVSD), PDA, or multiple VSDs.
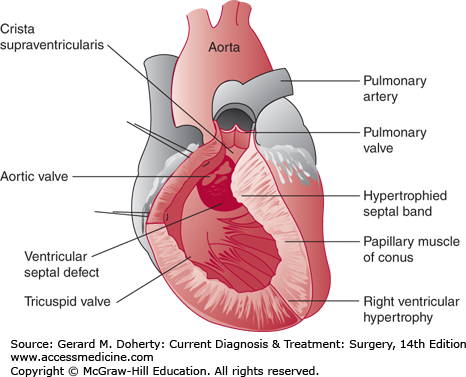
Patients with TOF develop cyanosis due to right-to-left shunting across the VSD. The degree of cyanosis depends on the severity of obstruction of the RV outflow tract. Frequently, cyanosis is mild at birth and may remain undetected for weeks or months. Neonates with severe infundibular obstruction or pulmonary atresia will develop symptoms shortly after birth and require a prostaglandin infusion to maintain ductal patency to ensure adequate pulmonary blood flow. In other patients, the RV outflow tract obstruction is minor, and the predominant physiology is that of a large VSD with left-to-right shunting and congestive heart failure.
The occurrence of intermittent cyanotic spells is a well-known feature of tetralogy. The etiology of “spelling” is still controversial but is clearly related to a transient imbalance between pulmonary and systemic blood flow. A spell may be triggered by hypovolemia or peripheral vasodilation (eg, after a bath or vigorous physical exertion). Spells may occur in neonates but are most frequently reported in infants between the ages of 3 and 18 months. Most spells resolve spontaneously within a few minutes, but some spells may be fatal. Older children have been observed to spontaneously squat to terminate spells. The squatting position is thought to increase systemic vascular resistance, which thereby favors pulmonary blood flow.
Cyanosis is the most frequent physical finding in TOF. Auscultation reveals a normal first heart sound and a single second heart sound. A systolic ejection murmur is present at the left upper sternal border. Older children may develop clubbing of the fingers and toes. Chest radiography typically demonstrates a boot-shaped heart due to elevation of the cardiac apex from RV hypertrophy. Pulmonary vascular markings are usually reduced. A right aortic arch may be present. An electrocardiogram shows RV hypertrophy. Echocardiography is definitive, and catheterization is not necessary in most cases.
The medical management of TOF is directed toward the treatment and prevention of cyanotic spells. The immediate treatment of the spelling patient includes administration of oxygen, narcotics for sedation, and correction of acidosis. Transfusion is indicated for anemic infants. Alpha-agonists are useful for increasing systemic vascular resistance (which favors pulmonary blood flow). Some centers have used beta-blockers as a form of long-term therapy to suppress the incidence of spells. Long-term complications of untreated TOF include clubbing of the fingers and toes, severe dyspnea on exertion, brain abscesses (secondary to right-to-left shunting), paradoxical embolization, and polycythemia (which may lead to cerebral thrombosis). Long-term survival is unlikely for most patients with untreated TOF.
All patients with TOF should undergo surgical repair. Asymptomatic patients should be repaired electively between 4 and 6 months of age. Early repair is indicated for neonates with severe cyanosis and for infants who have had a documented spell or worsening cyanosis.
Classically, the repair of TOF was accomplished in two stages. During the first stage, pulmonary blood flow was augmented by creating a connection (or shunt) between a systemic artery and the pulmonary artery. At the second stage, the shunt was taken down, and a complete repair was performed. The first shunt procedure was the Blalock-Taussig shunt, in which the subclavian artery was mobilized and divided distally, and an end-to-side anastomosis was created between the inferiorly deflected subclavian and the ipsilateral pulmonary artery. The modified Blalock-Taussig shunt is the most common type of shunt used today and consists of an interposition graft (polytetrafluoroethylene) between the innominate or subclavian artery and the ipsilateral pulmonary artery. Creation of a shunt may be accomplished with or without the use of cardiopulmonary bypass.
Currently, one-stage repair of TOF is preferred by most centers. Initial palliation with a shunt is still indicated for some patients who present a high risk for complete repair, such as those with multiple congenital anomalies, significant prematurity, severe concurrent illness, or an anomalous coronary artery crossing a hypoplastic infundibulum.
Complete repair of TOF is performed using a median sternotomy and cardiopulmonary bypass with bicaval venous cannulation. By a transatrial approach, the RV outflow tract can be examined through the tricuspid valve. Muscle bundles obstructing the RV outflow tract are divided or resected. The VSD is closed with a patch. Pulmonary valvotomy is performed, when indicated, via a vertical incision in the main pulmonary artery. When the pulmonary valve annulus or infundibulum is severely hypoplastic, a transannular outflow tract patch may be necessary to relieve the obstruction. When an anomalous coronary artery crosses the infundibulum, a transannular incision may be contraindicated. In these cases, and in patients with pulmonary atresia, placement of a conduit (cryopreserved homograft or bioprosthetic heterograft) between the right ventricle (via a separate ventriculotomy) and main pulmonary artery will be necessary. Patients who undergo construction of a transannular patch develop pulmonary insufficiency as a consequence. This is surprisingly well tolerated in most infants, as long as the tricuspid valve is competent.
As these patients grow older, some will develop RV failure due to chronic pulmonary insufficiency, and pulmonary valve replacement may be necessary. Currently, pulmonary valve replacement is indicated for symptomatic patients or those at risk of life-threatening arrhythmias. For asymptomatic patients, it is generally agreed that pulmonary valve replacement should be undertaken before irreversible RV dysfunction occurs. cMRI has evolved as a valuable diagnostic tool for evaluating RV size, function, anatomic detail, and other hemodynamic parameters. An RV size “cutoff” is debated, but recent recommendations suggest considering valve replacement before RV end-diastolic volume index exceeds approximately 160 mL/m2. Catheter-based pulmonary valve replacement is an option for some patients depending on the RV outflow tract anatomy.
The early mortality following repair of TOF is 1%-5%. The results are worse for patients with TOF and pulmonary atresia. Long-term complications include recurrent obstruction of the RV outflow tract and development of RV dysfunction due to chronic pulmonary insufficiency. Actuarial survival at 20 years is 90% with excellent functional status.
[PubMed: 22669402].
TGA is a congenital cardiac anomaly in which the aorta arises from the right ventricle and the pulmonary artery originates from the left ventricle (Figure 19–28). TGA is divided into dextro-looped (d-TGA) and levo-looped (l-TGA). The looping refers to the right or left looping of the primitive heart tube during fetal development, which determines whether the atria and ventricles are concordant (right atrium attaches to the right ventricle and left atrium attaches to the left ventricle) or discordant. l-TGA is associated with atrioventricular (AV) discordance (right atrium attaches to the left ventricle and left atrium attaches to the right ventricle) and is also termed congenitally corrected TGA. l-TGA is a rare variant of TGA and is beyond the scope of this chapter, which focuses on d-TGA. The defect can be subdivided into d-TGA with intact ventricular septum (IVS) (55%-60%) and d-TGA with VSD (40%-45%), one-third of which are hemodynamically insignificant. Pulmonic stenosis, causing significant left ventricular outflow tract obstruction, occurs rarely with an IVS and in approximately 10% of d-TGA/VSD.
Figure 19–28.
Typical transposition of the great arteries. The aorta arises from the morphologic right ventricle and is anterior and slightly to the right of the pulmonary artery, which originates from the morphologic left ventricle. Inset at bottom illustrates the independent systemic and pulmonary circulations, which may be connected by a patent ductus arteriosus or atrial septal defect. Inset at top illustrates a common relationship of the two great arteries in typical transposition.
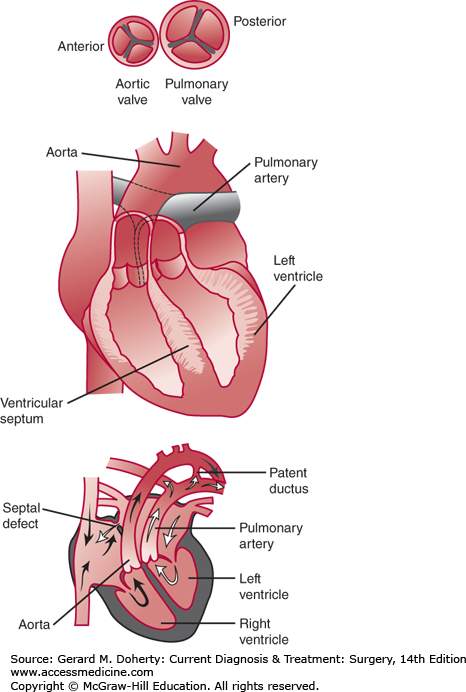
d-TGA is a relatively common cardiac anomaly and is the most common form of congenital heart disease presenting as cyanosis in the first week of life. The malformation accounts for approximately 10% of all congenital cardiovascular malformations in infants. The degree of cyanosis depends on the amount of mixing between the pulmonary and systemic circulations. In d-TGA, oxygenated pulmonary venous blood is returned to the lungs, and desaturated systemic blood is returned to the body. Because the two circulations exist in parallel, some mixing between them must occur to allow oxygenated blood to reach the systemic circulation and the desaturated blood to reach the lungs. Mixing may occur at a number of levels, most commonly at the atrial level through an ASD or a patent foramen ovale (PFO). Generally, two levels of mixing are necessary to maintain adequate systemic oxygen delivery with a VSD or PDA serving as an additional site for cardiac mixing. In d-TGA, there can be no fixed shunt in one direction without an equal amount of blood passing in the other direction; otherwise, one circulation would eventually empty into the other. Therefore, the amount of desaturated blood reaching the lungs (effective pulmonary blood flow) must equal the amount of saturated blood reaching the aorta (effective systemic blood flow). Clinical characteristics are dependent on the degree of mixing and the amount of pulmonary blood flow. These factors relate to the specific anatomic subtype of d-TGA. Neonates with d-TGA/IVS (or small VSD) have mixing limited to the atrial level and PDA. The ASD may be restrictive, and the PDA generally will close over the first days to week of life. As the degree of mixing decreases, the patient becomes increasingly cyanotic and will eventually suffer cardiovascular collapse. Fortunately, the majority of these neonates will manifest cyanosis early in life, which is recognized by a nurse or physician within the first hour in 56% and in the first day in 92%. In d-TGA with a large VSD, there is additional opportunity for mixing and increased pulmonary blood flow. The neonate with d-TGA/VSD may manifest only mild cyanosis, which may be initially overlooked. However, generally within 2-6 weeks, signs and symptoms of congestive heart failure will emerge. Tachypnea and tachycardia become prominent, while cyanosis may remain mild. Auscultatory findings are consistent with congestive heart failure with increased pulmonary blood flow, including a pansystolic murmur, third heart sound, mid-diastolic rumble, gallop, and narrowly split second heart sound with an increased pulmonary component. Neonates with d-TGA and significant pulmonary stenosis present with severe cyanosis at birth. Lesser degrees of pulmonary stenosis will result in varying levels of cyanosis.
In cases of d-TGA, the electrocardiogram is normal at birth, demonstrating the typical pattern of RV dominance. Although the classic chest radiographic appearance of an egg-shaped heart with a narrow superior mediastinum may be seen, this finding is often obscured by an enlarged thymic shadow. The abnormal ventriculoarterial connection is clearly seen on echocardiography, which demonstrates that the posterior great vessel arising from the left ventricle is a pulmonary artery that bifurcates soon after its origin. The anterior great vessel is the aorta and arises from the RV. Associated lesions, including VSD, left ventricular outflow tract obstruction, and coarctation, may also be diagnosed. Although used less frequently for diagnosis, cardiac catheterization may be helpful to improve cardiac mixing by means of balloon atrial septostomy.
The infant with d-TGA and severe cyanosis requires prompt diagnosis and treatment to improve mixing and increase the arterial oxygen saturation. The first intervention to improve mixing in a cyanotic newborn suspected of having TGA is to insure ductal patency by beginning an infusion of prostaglandin E1. In the presence of a restrictive ASD, a balloon atrial septostomy, a technique developed by William Rashkind in 1966, is performed. The procedure involves inserting a balloon-tipped catheter across the foramen ovale into the left atrium. Inflation and forcible withdrawal of the catheter tears the septum primum and enlarges the ASD. Mixing generally increases immediately, with a substantial increase in arterial oxygen saturation. Without intervention, d-TGA is universally fatal. Untreated, 30% of neonates will die in the first week of life, 50% by the first month, 70% within 6 months, and 90% by 1 year. The definitive surgical treatment of patients with d-TGA has changed dramatically with the advent of the arterial switch operation (ASO). The ASO, first successfully performed by Jatene in 1975, has become the optimal surgical procedure for infants with this condition. Current techniques have reduced the operative mortality to 2%-4%. The operative technique involves transection of both great vessels and direct reanastomosis to reestablish ventriculoarterial concordance. Additionally, the coronary arteries are removed from the anterior aorta and relocated to the posterior great vessel (neoaorta). The extensive experience gained with this procedure has confirmed that any variant of coronary artery anatomy can be successfully repaired, although certain unusual forms impose a higher risk. Because many patients with d-TGA have an IVS, left ventricular pressure falls early in life as pulmonary vascular resistance decreases. In this situation, it is essential that the arterial repair be performed within the first 2-3 weeks of life, while the left ventricle is still able to meet systemic workloads. In patients presenting later, the left ventricle can be retrained with a preliminary pulmonary artery band and aortopulmonary shunt followed by the definitive arterial repair. Although patients with large VSDs do not require early repair because of their decreased left ventricular pressure, experience has indicated that even in this subgroup, the operation is best performed within the first month of life, before secondary complications such as pulmonary hypertension, congestive heart failure, or infection develop.
Patients with fixed left ventricular outflow tract obstruction are not candidates for the arterial repair because correction would result in systemic ventricular outflow tract obstruction. Most of these patients also have large VSDs. Palliation early in life with systemic-to-pulmonary artery shunting is an option, with definitive repair postponed until somatic growth results in cyanosis as the shunt is outgrown. At that time, the Rastelli procedure is performed, in which left ventricular blood is redirected through the VSD to the anterior aorta by placement of an intraventricular patch. The pulmonary artery is ligated, and right ventricle-to-distal pulmonary artery continuity is reestablished with a valve-bearing conduit. An increasing number of experienced centers currently recommend early complete repair in the neonatal period using a Rastelli procedure. Early repair eliminates the interim morbidity and mortality associated with a systemic-to-pulmonary artery shunt and chronic cyanosis.
Current hospital survival for the ASO is 96.6%-97.2%. Generally, dTGA/IVS has had a lower mortality than d-TGA/VSD or d-TGA/VSD/PS. Hospital mortality for d-TGA/IVS is 2.2%, compared to 4.3% for d-TGA/VSD. Long-term survivals at 5-10 years and 15-20 years are 92.2%-97.9% and 91.6%-96%, respectively. The most common cause for reintervention is supravalvar pulmonary stenosis, occurring in 19.7%-30.3%. A recent study of 40 patients undergoing a Rastelli operation over a 20-year period revealed a hospital mortality of 0%, with Kaplan–Meier survival of 93% at 5, 10, and 20 years. Freedom from conduit replacement was 86% at 5 years and 59% at 20 years.
Truncus arteriosus is a rare anomaly that accounts for 0.4%-4% of all cases of congenital heart disease. A single arterial vessel arises from the heart, overriding the ventricular septum, and gives rise to the systemic, coronary, and pulmonary circulations. The Collett and Edwards classification of truncus arteriosus focuses on the origin of the pulmonary arteries from the common arterial trunk, as follows:
Type I: Common arterial trunk gives rise to a main pulmonary artery and the aorta.
Type II: Right and left pulmonary arteries arise directly from and in close proximity to the posterior wall of the truncus.
Type III: Right and left pulmonary arteries arise from more widely separated orifices on the posterior truncal wall.
Type IV: Branch pulmonary arteries are absent. Pulmonary blood flow is derived from aortopulmonary collaterals.
Persistent truncus arteriosus is the result of failed development of the aortopulmonary septum and subpulmonary infundibulum (conal septum). Normal septation leads to the development of both pulmonary and systemic outflow tracts, division of the semilunar valves, and formation of the aorta and pulmonary arteries. Failure of septation results in a VSD (absence of the infundibular septum), a single semilunar valve, and a single arterial trunk. Most cases are associated with a VSD with the superior margin of the defect formed by the truncal valve. The truncal valve leaflets are generally dysmorphic and their motion may be restricted. Leaflet number is highly variable, with about 65% tricuspid, 22% quadricuspid, 9% bicuspid, and rarely unicuspid or pentacuspid. As a result of these abnormally developed valve leaflets, a moderate or greater degree of truncal insufficiency is present in 20%-26% of patients. The pulmonary arteries are usually of normal size and most often arise from the left posterolateral aspect of the truncal artery, often in close proximity to the truncal valve and ostium of the left coronary artery.
Other cardiac anomalies are common and include an ASD (9%-20%), an interrupted aortic arch (10%-20%), and coronary ostial abnormalities (37%-49%) with the left coronary artery frequently noted to have a high origin, not uncommonly near the takeoff of the pulmonary arteries. Extracardiac anomalies are reported in approximately 28% of patients with truncus arteriosus. Described abnormalities include skeletal, genitourinary, gastrointestinal, and DiGeorge syndrome (11%).
The anatomy of truncus arteriosus results in obligatory mixing of systemic and pulmonary venous blood at the level of the VSD and truncal valve, which produces arterial saturations of 85%-90%. The systemic arterial saturation depends on the volume of pulmonary blood flow, which in turn is determined by the pulmonary vascular resistance (PVR). As the resistance begins to fall, excessive pulmonary circulation ensues and leads to pulmonary congestion and signs and symptoms of congestive heart failure. This nonrestrictive left-to-right shunt may cause early development of irreversible pulmonary vascular obstructive disease.
The presence of truncal valve abnormalities poses further hemodynamic burdens. Truncal valve regurgitation leads to ventricular dilation and low diastolic coronary perfusion pressures that can result in myocardial ischemia. Truncal valve stenosis promotes ventricular hypertrophy, increases the myocardial oxygen demand, and limits coronary and systemic perfusion, especially with the large volume of runoff into the pulmonary vascular bed.
Neonates with truncus arteriosus present with signs of congestive heart failure and collapsing peripheral pulses. Chest radiography shows marked cardiomegaly, pulmonary plethora, often with minimal thymus shadow, and a right aortic arch. The electrocardiogram most often depicts biventricular hypertrophy. Echocardiography is the diagnostic procedure of choice and can demonstrate the truncal vessel, the structure and function of the truncal valve, associated lesions such as interrupted aortic arch, and often the pulmonary arterial anatomy. Cardiac catheterization is not performed unless the anatomy is unclear, further information is needed about the status of the truncal valve, or the status of the pulmonary vasculature is unclear (ie, infants older than 3 months at diagnosis).
The natural history of patients born with truncus arteriosus is early demise. Approximately 40% of infants are dead within 1 month, 70% by 3 months, and 90% by 1 year. Early death is caused by congestive heart failure. Survivors may do well for a period of time until the development of pulmonary vascular obstructive disease and Eisenmenger syndrome. The ultimate treatment of truncus arteriosus is surgical correction in the neonatal period. Medical treatment is palliative and directed toward controlling congestive heart failure with fluid restriction, diuretics, and afterload reduction. Complete repair entails separating the pulmonary arteries from the truncus, repairing the resulting defect in the aorta, closing the VSD, and restoring continuity of the RV outflow tract with an extracardiac conduit (Figure 19–29). Severe truncal valve regurgitation requires truncal valve repair or replacement. An associated interrupted aortic arch is repaired by constructing a primary end-to-end anastomosis of the distal ascending aorta with proximal augmentation if necessary.
Figure 19–29.
Type 1 truncus arteriosus. A: The main pulmonary artery arises from the truncus arteriosus downstream to the truncal valve. A ventricular septal defect is present. B: The main pulmonary artery is incised from the truncus. The ventricular septal defect is closed with a patch. A valved conduit is sutured to the anterior wall of the right ventricle and the distal pulmonary artery.

The results of truncus arteriosus repair have improved greatly during the last two decades. Before the importance of early operation to avoid irreversible pulmonary vascular disease was appreciated, patients underwent repair at most institutions at an average age of 2-5 years with high mortality rates. Current hospital mortality for the neonatal repair of truncus arteriosus ranges between 4.3% and 17%, with the majority of deaths occurring in complex truncus arteriosus or in truncus arteriosus with associated severe truncal valve regurgitation. All patients will ultimately require reoperation for replacement of the right ventricle to pulmonary artery conduit with only 30%-42% being free from reoperation at 10 years. Thirty-year survival is approximately 75%-83%.
Total anomalous pulmonary venous connection (TAPVC) is a relatively uncommon congenital defect representing approximately 2% of all congenital heart anomalies. TAPVC encompasses a group of anomalies in which the pulmonary veins connect directly to the systemic venous circulation via persistent splanchnic connections. This abnormality results from failed transfer, in the normal developmental sequence, of pulmonary venous drainage from the splanchnic plexus to the left atrium. The most common classification system consists of four types: supracardiac (type 1), cardiac (type 2), infracardiac (type 3), and mixed (Figure 19–30). Partial anomalous pulmonary venous connection defines patients in whom some but not all venous drainage enters the left atrium, while the remaining veins connect to one or more persistent splanchnic veins.
Figure 19–30.
Common types of total anomalous pulmonary venous connection. Type 1: The pulmonary veins connect to a persistent left vertical vein, the innominate vein, and the right superior vena cava. Type 2: The pulmonary veins connect to the coronary sinus and the right atrium. Type 3: The pulmonary veins connect to an anomalous descending vein, a portal vein or persistent ductus venosus, and eventually enter the inferior vena cava.
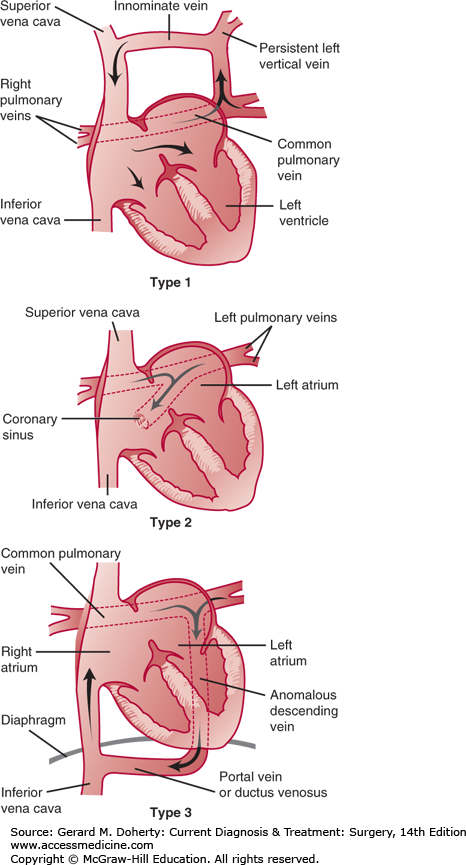
TAPVC can also be classified by the presence of obstruction. Impingement from surrounding structures or inadequate caliber of the draining pulmonary vein(s) can result in varying degrees of obstruction. Obstruction in supracardiac TAPVC can occur by compression of the ascending vertical vein between the left main stem bronchus and left pulmonary artery or by narrowing at the insertion of the vertical vein into the innominate vein. Obstruction is always present in the infracardiac type because the pulmonary venous blood must pass through the sinusoids of the liver. Obstruction is uncommon in the cardiac type.
Supracardiac occurs in approximately 45% of patients. The common pulmonary vein drains superiorly into the innominate vein, superior vena cava, or azygous vein via an ascending vertical vein. Cardiac TAPVC occurs in approximately 25% of patients. The pulmonary venous confluence drains into the coronary sinus or, on rare occasions, individual pulmonary veins will connect directly into the right atrium. Infracardiac TAPVC occurs in approximately 25% of patients. The pulmonary venous confluence drains into a descending vertical vein through the diaphragm into the portal vein or ductus venosus. Finally, a mixed type of TAPVC occurs in approximately 5% of patients and can involve any or all components of the previous three types.
TAPVC produces a mixing lesion because oxygenated blood from the pulmonary system drains back into the systemic venous circulation. The size of the ASD dictates the distribution of blood flow. Most patients with unobstructed TAPVC have few or no symptoms in infancy and present with signs and symptoms similar to an ASD. In the neonate with obstructed TAPVC, venous drainage from the pulmonary vasculature is impaired, leading to pulmonary venous hypertension and pulmonary edema. In severe cases, this increased pressure will lead to reflexive vasoconstriction of the pulmonary vasculature with pulmonary hypertension. Patients with obstruction present early in life with profound cyanosis from pulmonary edema.
The diagnosis can be made with echocardiographic identification of the anomalous connection of the pulmonary venous confluence to the systemic venous system. The ASD and other associated anomalies can be delineated. Cardiac catheterization is rarely necessary unless accurate measurement of PVR is needed.
The management of TAPVC is surgical repair. In patients with obstruction, medical management for stabilization may be employed but is often unsuccessful and should not delay surgical intervention.
The principle of operative repair is to establish an unobstructed communication between the pulmonary venous confluence and the left atrium, interrupt the connections with the systemic venous circulation, and close the ASD. The specific repair is dependent on the type of anomalous connection.
Supracardiac TAPVC The repair of supracardiac TAPVC may be performed with moderate hypothermia (28-32°C) and bicaval cannulation or with a brief period of hypothermic (18°C) circulatory arrest. The optimal approach is to retract the ascending aorta to the left and the superior vena cava to the right to expose the pulmonary venous confluence. This approach provides excellent exposure without distortion of the heart or venous structures. The vertical vein can be identified and ligated (just prior to opening the confluence) outside the pericardium at the level of the innominate vein. A transverse incision is made in the pulmonary venous confluence, and a parallel incision is placed in the dome of the left atrium beginning at the base of the left atrial appendage. The common pulmonary vein is then anastomosed to the left atrium, taking care to construct an unrestrictive connection. A right atriotomy is made to close the ASD.
Cardiac TAPVC The repair of cardiac TAPVC can be performed with bicaval cannulation and moderate hypothermia with the use of a vent or a cardiotomy sucker to capture the pulmonary venous return. A right atriotomy is performed with identification of the ASD and the orifice of the coronary sinus. The roof of the coronary sinus is excised into the left atrium. A patch of pericardium or prosthetic material is then placed to close the enlarged ASD, effectively channeling the pulmonary venous and coronary sinus return into the left atrium. The conduction system travels in proximity to the coronary sinus, and care must be taken while suturing the patch in this area to avoid heart block.
Infracardiac TAPVC For infracardiac connections, a brief period of hypothermic circulatory arrest is often required. The heart is rotated superiorly. The descending vertical vein is identified by opening the posterior pericardium. The connection to the descending vertical vein is ligated at the level of the diaphragm. An incision is made along the length of the pulmonary venous confluence with a parallel incision on the posterior wall of the left atrium. The pulmonary venous confluence is then anastomosed to the left atrium, taking care not to narrow the connection. Tissue from the descending vertical vein can be used in the anastomosis. A right atriotomy is performed through which the ASD is closed.
Recurrent pulmonary venous obstruction The approach to recurrent pulmonary venous obstruction is dependent on the level of obstruction. Obstruction can develop at the anastomosis or within the individual pulmonary veins. The latter may initially present as an anastomotic constriction, as the true extent of obstruction is not always apparent at first. Isolated narrowing of the anastomosis between the common pulmonary vein and left atrium often can be repaired with revision or patch augmentation of the anastomosis.
Obstruction of the individual pulmonary venous ostia is the greater challenge. Although the obstruction may initially appear to be limited to the ostium, progressive narrowing along the entire length of the vein into the hilum of the lung may occur with time. Repair of this lesion is technically challenging, and recurrent early obstruction is common. A new approach to recurrent pulmonary vein stenosis employs a “sutureless” technique utilizing in situ pericardium to create a neoatrium. The theory behind this repair is based on the concept that pulmonary venous obstruction results from inflammation induced locally by suture placement. Repair involves wide unroofing of the narrowed portion of each involved pulmonary vein from the left atrial anastomosis to the hilum. A wide flap of pericardium is then elevated with care taken to avoid disruption of posterior adhesions and injury to the phrenic nerve. This flap of pericardium is rotated over the unroofed pulmonary veins and sutured to the left atrial wall away from the venous ostia. A large neoatrium is then created into which pulmonary venous return can drain.
Early mortality in patients undergoing repair of TAPVC is associated with the initial degree of obstruction present. Early diagnosis and repair as well as optimal postoperative management, including aggressive treatment of pulmonary hypertension, have resulted in a dramatic reduction in operative risk. For patients surviving the perioperative period, the long-term survival and functional status are excellent.
Recurrent venous obstruction develops in 5%-17.5% of patients. Results following balloon angioplasty and/or stent insertion have been disappointing, and recurrent stenoses are the rule. Individual patch angioplasty of the ostia has also been utilized with poor long-term results. Lung transplantation has been considered in severe cases of extensive, bilateral disease. The mortality associated with reoperation for obstruction can be up to 59% when bilateral stenoses are present. The use of the sutureless technique for recurrent pulmonary vein obstruction has demonstrated improved survival and decreased recurrence.
[PubMed: 22797519].
[PubMed: 22892140].
[PubMed: 23054615].
Tricuspid atresia refers to single-ventricle hearts that lack a communication between the right atrium and right ventricle. The only outlet for the right atrium is the ASD. If present, aneurysmal tissue of the septum primum may prolapse into the left atrium. The left atrium is normal in morphology but is often dilated. A normal communication with a mitral valve between the left atrium and left ventricle is present. The right ventricle is diminutive in size without an inlet portion. It is connected to the left ventricle by a VSD surrounded completely by muscle. The anatomic subtypes of tricuspid atresia are based on the relationship of the great arteries. Type I defects (70%) have normally related great vessels, type II (30%) have transposed great arteries, and type III (rare) have congenitally corrected TGAs. These types are further subclassified by the degree of obstruction to pulmonary blood flow, which is present in 45%-75% of patients. Aortic valve (10%) and aortic arch (25%) obstruction may also be present. Patients with tricuspid atresia are at increased risk for Wolff–Parkinson–White syndrome due to congenital and surgically acquired pathways.
The clinical presentation depends on the relationship of the great vessels and degree of restriction at the level of the atrial and ventricular septum. Most infants present with some degree of cyanosis. Systemic output is usually unobstructed. Prostaglandins may be necessary to maintain ductal patency in infants with severe obstruction to pulmonary blood flow.
The initial palliation for most patients is the placement of a modified Blalock-Taussig shunt to maintain adequate pulmonary blood flow. A more complex initial palliation with a Norwood procedure may be necessary in the case of transposed great vessels. The remainder of the palliation involves a hemi-Fontan and Fontan procedure (discussed in the section on Hypoplastic Left Heart Syndrome).
The overall survival for tricuspid atresia is similar to that reported for other single-ventricle lesions palliated with a Fontan procedure. The survival is 83% at 1 year, 70% at 10 years, and 60% at 20 years.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


