Chapter 38 Hypothalamic, pituitary and sex hormones
• Many of the pituitary hormones and their hypothalamic releasing factors are used in diagnosis or therapy.
• The main therapeutic use of pituitary hormones is of growth hormone (anterior pituitary) and those from the posterior pituitary: oxytocin and vasopressin.
• Vasopressin (antidiuretic hormone) is used both for its vasoconstrictor effect (in the treatment of oesophageal varices) and for its antidiuretic action.
• The main hypothalamus–pituitary target organ axis for therapeutic intervention is that controlling reproductive hormones, especially in women.
• Suppression of oestrogen and/or androgen production is used in the treatment of tumours stimulated by these: breast and prostate.
• Therapy in women is used to suppress ovulation (contraceptives), to stimulate ovulation (fertility treatment) or to mimic ovarian endocrine function (post-menopausal hormone replacement therapy, HRT).
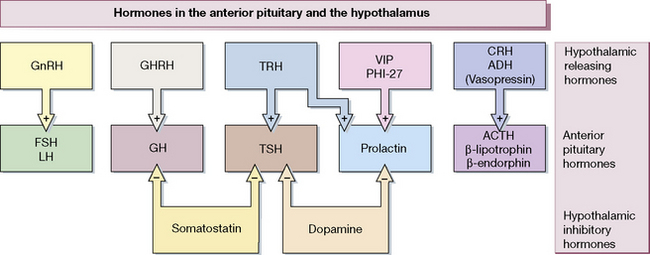
Figure 38.1 shows the hypothalamo-pituitary axes. The hypothalamus and pituitary glands form the centre of the ‘endocrine orchestra’. We will here describe the hypothalamic releasing hormones, and the anterior and posterior pituitary gland hormones, and drugs that are used to manipulate these axes.
These hormones, analogues (agonists) and antagonists can be used:
• To analyse the functional integrity of endocrine control systems.
• As replacement in hormone deficiency states.
• To modify malfunction of endocrine systems.
• To alter normal function where this is inconvenient, e.g. contraception.
The scope of the specialist endocrinologist continues to increase in amount and in complexity and only an outline is appropriate here.
Hypothalamic and anterior pituitary hormones
The hypothalamus releases a number of locally active hormones that stimulate or inhibit pituitary hormone release (see Fig. 38.1).
Octreotide
is a synthetic analogue of somatostatin having a longer action (t½ 1.5 h). It is administered subcutaneously two or three times daily; a depot formulation is available for deep intramuscular injection once a month. Lanreotide is much longer acting than octreotide, and is administered intramuscularly twice a month. Uses of the somatostatin analogues include acromegaly, carcinoid (serotonin-secreting) tumours and other rare tumours of the alimentary tract. An unlicensed use of octeotride is the termination of variceal bleeding (see p. 548). Radiolabelled somatostatin is used to localise, and in higher doses to treat, metastases from neuroendocrine tumours which often bear somatostatin receptors. Both octreotide and lanreotide are now available as a monthly slow-release preparation.
Growth hormone, somatrophin
Growth hormone is approved for treatment of children with short stature due not just to growth hormone deficiency, but also to Turner’s syndrome, renal failure, small size for gestational age, Prader–Willi syndrome1 and, most recently, idiopathic short stature. Treatment is continued until closure of the epiphyses. Subsequent treatment into adulthood is also warranted where UK National Institute for Health and Clinical Excellence (NICE) guidelines are fullfilled. Growth hormone therapy should be confined to specialist clinics.
Posterior pituitary hormones and analogues
Desmopressin
Nephrogenic diabetes insipidus, as is to be expected, does not respond to antidiuretic hormone.
In bleeding oesophageal varices, use is made of the vasoconstrictor effect of vasopressin (as terlipressin, a vasopressin prodrug); see page 564.
In haemophilia, desmopressin can enhance blood concentration of factor VIII.
Felypressin is used as a vasoconstrictor with local anaesthetics.
Diabetes insipidus: vasopressin deficiency
Sex (gonadal) hormones and antagonists: steroid hormones
Androgens
Preparations and choice of androgens
Testosterone given orally is subject to extensive hepatic first-pass metabolism (see p. 86) and it is therefore usually given by other routes. Androgens are available for oral, buccal, transdermal or depot administration.
Transdermal preparations
Non-scrotal patches are applied to the skin of the upper arms, back, abdomen and thighs.
Antiandrogens (androgen antagonists)
Cyproterone
• Competition with testosterone for receptors in target peripheral organs (but not causing feminisation as do oestrogens); it reduces spermatogenesis even to the level of azoospermia (reversing over about 4 months after the drug is discontinued); abnormal sperm occurs during treatment.
• Competition with testosterone in the CNS, reducing sexual drive and thoughts, and causing impotence.
• Some agonist progestogenic activity on hypothalamic receptors, inhibiting gonadotrophin secretion, which also inhibits testicular androgen production.
Uses
Finasteride and dutasteride (see p. 462), which inhibit conversion of testosterone to dihydrotestosterone, have localised antiandrogen activity in tissues where dihydrotestosterone is the principal androgen; they are therefore useful drugs in the treatment of benign prostatic hypertrophy.
Spironolactone (see p. 563) also has antiandrogen activity and may help hirsutism in women (as an incidental benefit to its diuretic effect). Androgen secretion may be diminished by continued use of a gonadorelin (LHRH) analogue (see p. 597).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


