FIGURE 53-1 ECG in a patient with HCM. Note prominent QRS voltages and deep T-wave inversions.
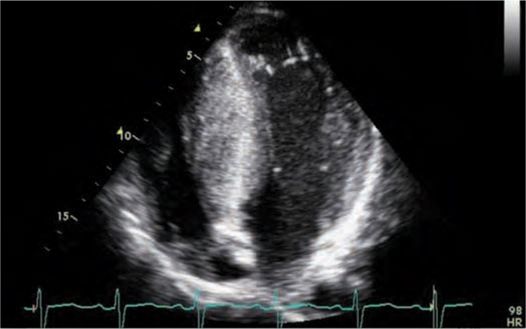
FIGURE 53-2 Echocardiogram demonstrating asymmetric hypertrophy with nearly a 5-cm septum.
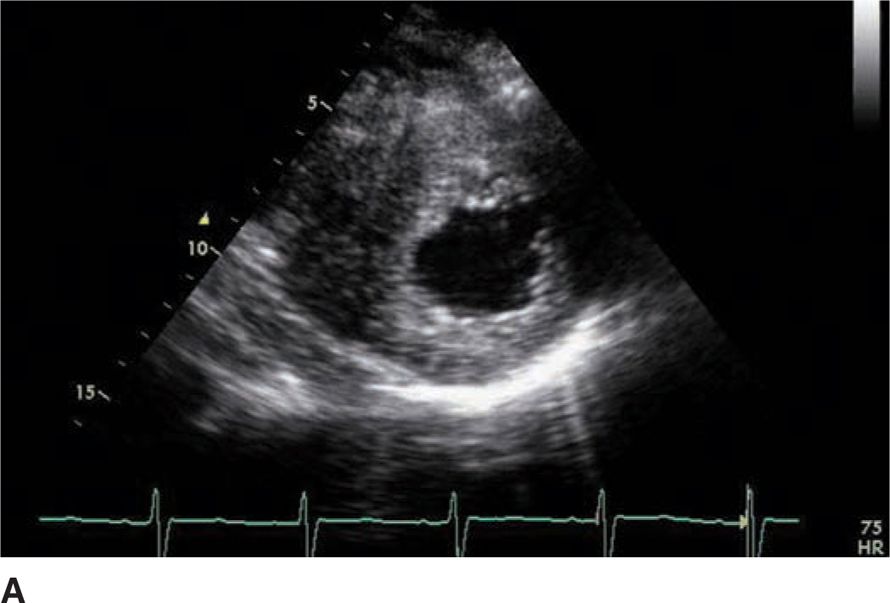
FIGURE 53-3A Parasternal short axis view of left ventricle in diastole.
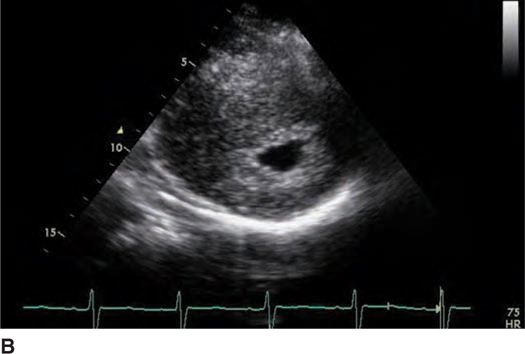
FIGURE 53-3B Parasternal short axis view of the left ventricle in systole.
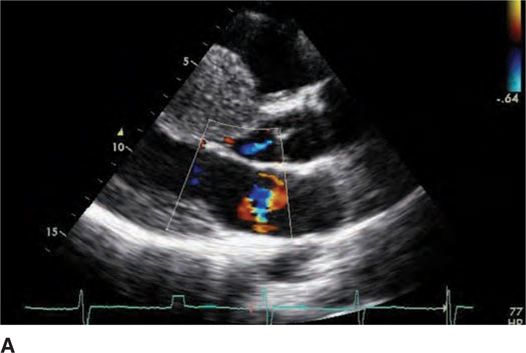
FIGURE 53-4A Parasternal long-axis view with color Doppler in diastole demonstrating normal opening of mitral valve.
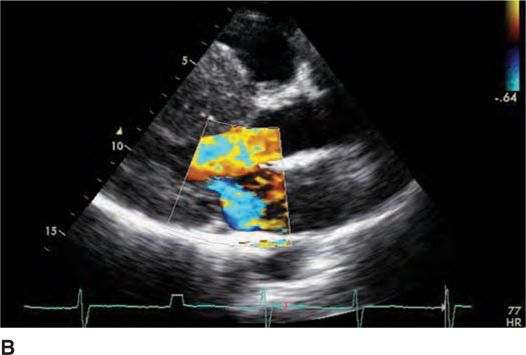
FIGURE 53-4B Parasternal long-axis view during systole with color Doppler demonstrating increased turbulent flow through the aortic valve with SAM and resulting mitral regurgitation.
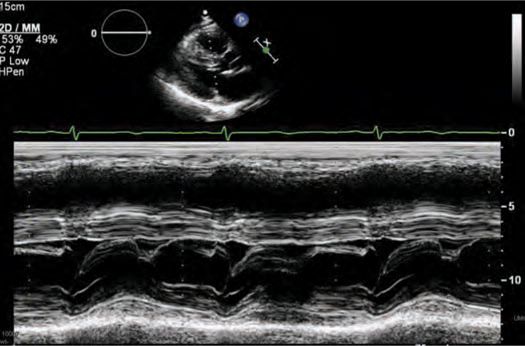
FIGURE 53-5 M-mode across the mitral valve demonstrating SAM with mitral-septal contact.
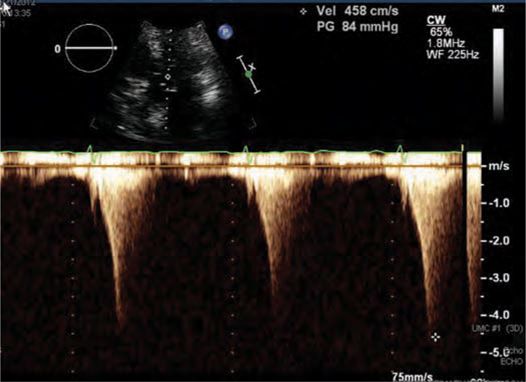
FIGURE 53-6 LVOT gradient after provocation with peak gradient recorded as 84 mm Hg.
The patient was started on metoprolol tartrate 50 mg twice daily and rapidly up titrated to 200 mg twice daily. Genetic counseling and genetic testing were performed, revealing a β-myosin heavy chain mutation and confirming the diagnosis of hypertrophic cardiomyopathy (HCM).
Based on a family history of unexplained sudden death and an LV wall thickness of greater than 3 cm, he underwent implantation of an ICD without complication for prevention of sudden cardiac death (SCD). Unfortunately, he had no improvement in symptoms despite high dose metoprolol and initiation of disopyramide. After 3 months of medical therapy and an attempt at pacing the right ventricle with the already implanted ICD, the LVOT gradient with provocation remained >50 mm Hg. Given his young age and refractory NYHA class III symptoms, he was referred to for myectomy. This operation was performed without complication and resulted in immediate improvement of symptoms. At follow-up 1 year later, the patient remains symptom-free with no resting or provocable LVOT gradient and no recorded arrhythmias.
EPIDEMIOLOGY
Inherited as an autosomal-dominant disorder, HCM is variably penetrant resulting in striking variations in phenotype even among first-degree relatives. Traditionally, the prevalence of HCM has been cited as 1:500, but due to the lack of symptoms in many individuals who may carry the genetic trait, the true prevalence may be much higher.1,2
ETIOLOGY AND PATHOPHYSIOLOGY
Clinically, hypertrophic cardiomyopathy (HCM) is generally described as left ventricular hypertrophy (LVH) with nondilated ventricular chambers in the absence of another cardiac or systemic disease that would be capable of producing the same degree of hypertrophy.1,2 Patients may harbor nearly any diffuse or segmental pattern of hypertrophy and fibrosis with presentations ranging from completely asymptomatic to NYHA class II-IV heart failure to SCD.
The onset of symptoms and recognition of the disease may occur at any age from infancy to adulthood, but commonly develops during periods of accelerated body growth, such as adolescence. For that reason clinical screening is generally recommended in patients with genetically proven HCM beginning at age 12 and continuing yearly until age 18 to 21 followed by screening every 5 years thereafter.1
The availability of genetic testing has aided in the identification of an increasing number of genotype-positive, phenotype-negative patients. Indeed, there are now well over 1400 documented mutations that, when coupled with a sensitive genetic substrate and environmental triggering factors, are responsible for the variety of clinical presentations of HCM.1,3 The genetic mutations allow the discrimination of HCM patients from patients with other genetic causes of LVH, such as Fabry disease, who would require much different management.
The original associations of genetic variants to HCM patients were isolated to myofilament (sarcomeric) proteins with the most common mutations occurring in the β-myosin heavy chain and cardiac myosin-binding protein C.3 Recent investigations have focused not only on the sarcomeric proteins but on the associated contractile apparatus, as well, with new disease causing mutations classified as Z-disc HCM and calcium-handling HCM.3 Ongoing investigations are trying to associate disease outcomes with various mutations and/or the affected protein subunits.1,3 The cellular alterations caused by these mutations lead to varying degrees of hypertrophy and fibrosis in the heart. This in turn may eventually lead to systolic anterior motion of the mitral valve with LVOT obstruction; the resulting symptoms of dyspnea, congestive heart failure, and syncope; and the risk of cardiac arrhythmias including atrial fibrillation, ventricular tachycardia, and ventricular fibrillation.
DIAGNOSIS
Initial clinical evaluation of a patient includes a thorough personal and family history to elicit symptoms of syncope, exercise-induced chest discomfort, dyspnea, palpitations, and/or family history of SCD. The physical examination should include a careful auscultation of the patient with appropriate maneuvers such as stand-squat-stand to evaluate the characteristics of the murmur.
Initial screening should include each of the following:
• Transthoracic echocardiogram
• 24-hour Holter monitoring
• Genetic assays if feasible
• Graded exercise test
The 12-lead ECG (see Figure 53-1) should demonstrate LVH and repolarization abnormalities. Incongruence between the ECG and echo findings should lead to definitive evaluation with either cardiac magnetic resonance imaging (CMRI)(Figure 53-7A and 53-7B) or invasive hemodynamic studies in the cath lab. In order to help assess the risk of SCD, HCM patients should also have 24-hour Holter monitoring and a graded exercise test performed at the time of diagnosis to evaluate for the presence of asymptomatic arrhythmias and determine the blood pressure response during exercise.1,4
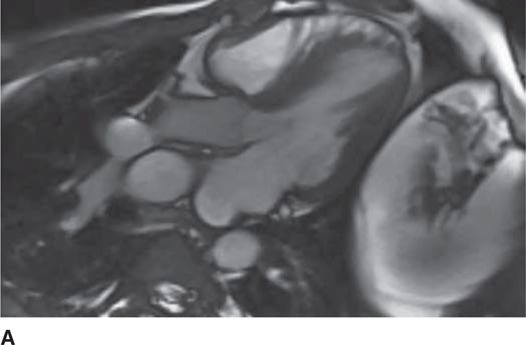
FIGURE 53-7A Cardiac MR with open mitral inflow during diastole. (Used with permission of Dr. Andrew Rivard at University of Mississippi Medical Center, Jackson, MS.)
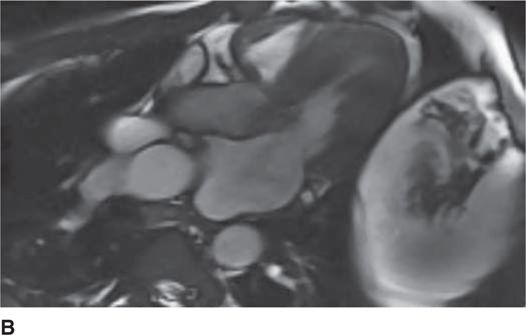
FIGURE 53-7B Cardiac MR with obstruction of the LVOT during systole due to SAM of the mitral valve. (Used with permission of Dr. Andrew Rivard at University of Mississippi Medical Center, Jackson, MS.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


