Chapter 26 Hyperlipidaemias
Some pathophysiology
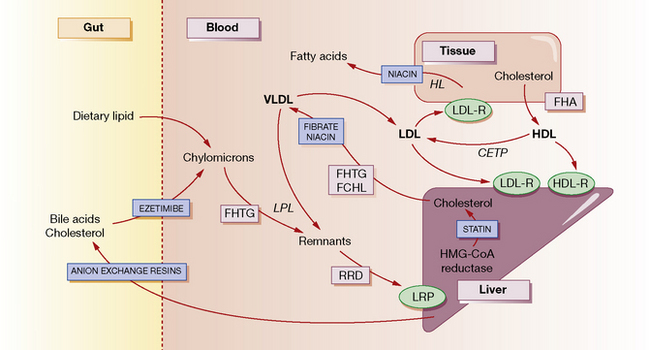
The normal function of lipoproteins is to distribute and recycle cholesterol. The pathways of lipid metabolism and transport and their primary (inherited) disorders are shown in Figure 26.1, and can be summarised thus:
• Cholesterol is absorbed from the intestine within chylomicrons. These are catabolised by lipoprotein lipase (LPL) to remnants, which are taken up by the hepatic low-density lipoprotein (LDL) receptor-related protein (LRP). A specific active transport mechanism also carries cholesterol across the gut mucosa (see below, ezetimibe).
• Cholesterol is synthesised de novo within the liver and peripheral tissues where, for example, it is converted to steroid hormones or incorporated into cell membranes. Much of the hepatic cholesterol enters the circulation as very-low-density lipoprotein (VLDL) and is metabolised to remnant lipoproteins after LPL removes triglyceride. The remnant lipoproteins are removed by the liver through apolipoprotein E receptors or LDL receptors (LDL-R), or further metabolised to LDL and then removed by peripheral tissues or the liver by LDL receptors.
• The quantity of cholesterol transported from the liver to peripheral tissues exceeds its catabolism there, and mechanisms exist to return about half of the cholesterol to the liver. Through this ‘reverse transport’, cholesterol is carried by high-density lipoprotein (HDL) from peripheral cells to the liver, where it is taken up by a process involving hepatic lipase. Cholesterol in the plasma is also recycled to LDL and VLDL by cholesterol-ester transport protein (CETP).
• Cholesterol in the liver is reassembled into lipoproteins, or secreted in bile and bile acids (essential for fat digestion and absorption), and then recycled by absorption or excreted in the faeces.
Lipid disorders
There are five primary inherited lipoprotein disorders that disturb lipid metabolism at the points indicated in Figure 26.1. These are:
• Familial hypertriglyceridaemia (FHTG) (rare), including lipoprotein lipase (LPL) deficiency, in which low LPL activity results in decreased removal, and thus increased levels of plasma triglyceride; there is increased hepatic secretion and thus a raised plasma concentration of triglyceride-rich VLDL. Patients are at risk of recurrent acute pancreatitis when plasma triglyceride exceeds 10 mmol/L, and especially when greater than 20 mmol/L.
• Familial combined hyperlipidaemia (FCHL) (common and most important) in which there is increased hepatic secretion of apolipoprotein B containing VLDL, and conversion to LDL; in consequence plasma LDL and VLDL levels are raised. Patients exhibit macrovascular disease (coronary heart, peripheral and cerebral).
• Remnant removal disease (RRD, also called remnant lipaemia, familial dysbetalipoproteinaemia) (uncommon) in which there is a defect of apolipoprotein E. This is the major ligand that allows internalisation and subsequent metabolism of remnant particles derived from VLDL and chylomicrons. The consequence is accumulation of VLDL remnants called intermediate-density lipoprotein (IDL), with cholesterol and triglycerides usually in the range 6–9 mmol/L. Patients experience severe macrovascular disease (as above).
• Familial hypoalphalipoproteinaemia (FHA, Tangier disease) (rare) in which the serum concentration of (protective) HDL is very low. Coronary heart and peripheral vascular disease result.
• Familial hypercholesterolaemia (FH) (common) is characterised by raised plasma levels of total and LDL cholesterol. It is caused by mutations in either the LDL receptor, Apo B-100 or PCSK9 genes. In the less severe heterozygous form, it affects about 1 in 500 of the population (who carry one copy of the mutant gene). LDL cholesterol levels are increased from childhood. Untreated, half the males will be dead by age 60 years, females 10 years later. The principal consequence is premature coronary heart disease, but occasionally also peripheral and cerebrovascular disease.
Sites of drug action
In general, drugs act to reduce the concentration of cholesterol within hepatocytes, producing a compensatory increase in LDL receptors on their surface, and increased uptake of cholesterol-rich LDL particles from the bloodstream (see Fig. 26.1). Statins decrease the synthesis of cholesterol and hence secretion of VLDL, and increase the surface expression of hepatic LDL receptors. Bile acid-binding (anion exchange) resins deplete the bile acid and thus the cholesterol pool. Fibrates decrease the secretion of VLDL and increase the activity of LPL, thereby increasing the removal of triglycerides. Nicotinic acid decreases fatty acid production in the tissues and the secretion of VLDL and clearance of HDL. It also enhances LPL. Ezetimibe blocks the uptake of cholesterol from the gut by targeting a specific cholesterol transporter.
Management
• Hyperlipidaemias are common; 66% of the adult UK population have a plasma cholesterol concentration in excess of 5.0 mmol/L, the lowest concentration generally associated with initiating drug treatment (in fact, statistical correlation with cardiovascular risk can be shown for cholesterol concentrations well below this value). The decision to treat an individual is made not just on the value for plasma lipids, but also on the absolute cardiovascular risk.
• Investigation of hyperlipidaemia must be directed initially at excluding contributory causes, i.e. secondary hyperlipidaemias (see above). None of these should be assumed to be the sole cause, even if present. Long-term decisions on management should be initiated only on the basis of at least two fasting blood samples.
• All patients (and their spouses/partners, if appropriate) should receive advice on lifestyle, diet and weight control, which are important components of overall macrovascular risk prevention. Dietary treatment of hypercholesterolaemia has a modest effect in the individual (at best an 8% reduction in LDL cholesterol is possible), but diet and weight reduction are more effective for hypertriglyceridaemia. Intake of total fat, especially saturated fat, should be reduced (and partially replaced with monounsaturated and polyunsaturated fats); spreads containing plant sterols and stanols, e.g. Benecol and Flora Proactiv, taken as part of a mixed meal are useful as they can reduce plasma cholesterol by up to 10%. Increasing attention is now paid the hydrogenated fat content of food (hydrogen bubbled through liquid oils improves texture, flavour and shelf-life). In some individuals, especially those with mixed hyperlipidaemia (raised cholesterol and triglyceride levels, often due to secondary factors on a polygenic background), successful adherence to dietary advice and weight loss produces very significant improvements. Patients with remnant lipaemia (RRD hyperlipidaemia) may respond excellently to diet and weight loss (and, possibly, the addition of a fibrate).
• Much of the work of lipid clinics is taken up with attending to multiple interacting risk factors such as hypertension, diabetes, thyroid disease and smoking, as well as to the lipid abnormality. The Joint British Societies Guidelines1 stress the importance of identifying the high-risk individuals who need intervention with diet and lifestyle changes and, if necessary, pharmacotherapy. Equal priority is given to: (1) patients with clinical atherosclerotic disease in any territory, (2) those with diabetes mellitus types 1 and 2 (the most numerous), and (3) those without symptomatic vascular disease but whose 10-year risk of developing it is greater than 20%.
• The decision to use lipid-lowering drugs is made on the basis of the overall absolute cardiovascular disease risk (CVD; see below and NICE guidance in footnote 1), e.g. evidence of existing CVD, hypertension, diabetes mellitus and a positive family history. This will often be quantified using an appropriate risk equation as the patient’s 10-year risk of developing cardiovascular disease: the threshold is set at 20% in the current NICE guidance1. The justification is easiest in two cases. Firstly, for primary prevention in the relatively small number of patients who are asymptomatic but have familial forms of hyperlipdaemia, e.g. patients with FH and remnant lipaemia. Secondly, for secondary prevention in patients who have evidence of CVD (previous myocardial infarction (MI), angina pectoris, stroke or transient ischaemic attack (TIA)), peripheral vascular disease or diabetes mellitus: in the landmark Scandinavian ‘4 S’ Study,2
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


