Chapter 8 High-throughput screening
Introduction: a historical and future perspective
Systematic drug research began about 100 years ago, when chemistry had reached a degree of maturity that allowed its principles and methods to be applied to problems outside the field, and when pharmacology had in turn become a well-defined scientific discipline. A key step was the introduction of the concept of selective affinity through the postulation of ‘chemoreceptors’ by Paul Ehrlich. He was the first to argue that differences in chemoreceptors between species may be exploited therapeutically. This was also the birth of chemotherapy. In 1907, Ehrlich identified compound number 606, Salvarsan (diaminodioxy-arsenobenzene) (Ehrlich and Bertheim, 1912), which was brought to the market in 1910 by Hoechst for the treatment of syphilis, and hailed as a miracle drug (Figure 8.1).

Today’s marketed drugs are believed to target a range of human biomolecules (see Chapters 6 and 7), ranging from various enzymes and transporters to G-protein-coupled receptors (GPCRs) and ion channels. At present the GPCRs are the predominant target family, and more than 800 of these biomolecules have been identified in the human genome (Kroeze et al., 2003). However, it is predicted that less than half are druggable and in reality proteases and kinases may offer greater potential as targets for pharmaceutical products (Russ and Lampel, 2005). Although the target portfolio of a pharmaceutical company can change from time to time, the newly chosen targets are still likely to belong to one of the main therapeutic target classes. The selection of targets and target families (see Chapter 6) plays a pivotal role in determining the success of today’s lead molecule discovery.
Over the last 15 years significant technological progress has been achieved in genomic sciences (Chapter 7), high-throughput medicinal chemistry (Chapter 9), cell-based assays and high-throughput screening. These have led to a ‘new’ concept in drug discovery whereby targets with therapeutic potential are incorporated into biochemical or cell-based assays which are exposed to large numbers of compounds, each representing a given chemical structure space. Massively parallel screening, called high-throughput screening (HTS), was first introduced by pharmaceutical companies in the early 1990s and is now employed routinely as the most widely applicable technology for identifying chemistry starting points for drug discovery programmes.
Nevertheless, HTS remains just one of a number of possible lead discovery strategies (see Chapters 6 and 9). In the best case it can provide an efficient way to obtain useful data on the biological activity of large numbers of test samples by using high-quality assays and high-quality chemical compounds. Today’s lead discovery departments are typically composed of the following units: (1) compound logistics; (2) assay development and screening (which may utilize automation); (3) tool (reagent) production; and (4) profiling. Whilst most HTS projects focus on the use of synthetic molecules typically within a molecular weight range of 250–600 Da, some companies are interested in exploring natural products and have dedicated research departments for this purpose. These groups work closely with the HTS groups to curate the natural products, which are typically stored as complex mixtures, and provide the necessary analytical skills to isolate the single active molecule.
Compared with initial volume driven HTS in the 1990s there is now much more focus on quality-oriented output. At first, screening throughput was the main emphasis, but it is now only one of many performance indicators. In the 1990s the primary concern of a company’s compound logistics group was to collect all its historic compound collections in sufficient quantities and of sufficient quality to file them by electronic systems, and store them in the most appropriate way in compound archives. This resulted in huge collections that range from several hundred thousand to a few million compounds. Today’s focus has shifted to the application of defined electronic or physical filters for compound selection before they are assembled into a library for testing. The result is a customized ensemble of either newly designed or historic compounds for use in screening, otherwise known as ‘cherry picking’. However, it is often the case that the HTS departments have sufficient infrastructure to enable routine screening of the entire compound collection and it is only where the assay is complex or relatively expensive that the time to create ‘cherry picked’, focused, compound sets is invested (Valler and Green, 2000).
In assay development there is a clear trend towards mechanistically driven high-quality assays that capture the relevant biochemistry (e.g. stochiometry, kinetics) or cell biology. Homogeneous assay principles, along with sensitive detection technologies, have enabled the miniaturization of assay formats producing a concomitant reduction of reagent usage and cost per data point. With this evolution of HTS formats it is becoming increasingly common to gain more than one set of information from the same assay well either through multiparametric analysis or multiplexing, e.g. cellular function response and toxicity (Beske and Goldbard, 2002; Hanson, 2006; Hallis et al., 2007). The drive for information rich data from HTS campaigns is no more evident than through the use of imaging technology to enable subcellular resolution, a methodology broadly termed high content screening (HCS). HCS assay platforms facilitate the study of intracellular pharmacology through spatiotemporal resolution, and the quantification of signalling and regulatory pathways. Such techniques increasingly use cells that are more phenotypically representative of disease states, so called disease-relevant cell lines (Clemons, 2004), in an effort to add further value to the information provided.
Screening departments in large pharmaceutical companies utilize automated screening platforms, which in the early days of HTS were large linear track systems, typically five metres or more in length. The more recent trends have been towards integrated networks of workstation-based instrumentation, typically arranged around the circumference of a static, rotating robotic arm, which offers greater flexibility and increased efficiency in throughput due to reduced plate transit times within the automated workcell. Typically, the screening unit of a large pharmaceutical company will generate tens of millions of single point determinations per year, with fully automated data acquisition and processing. Following primary screening, there has been an increased need for secondary/complementary screening to confirm the primary results, provide information on test compound specificity and selectivity and to refine these compounds further. Typical data formats include half-maximal concentrations at which a compound causes a defined modulatory effect in functional assays, or binding/inhibitory constants. Post-HTS, broader selectivity profiling may be required, for active compounds against panels of related target families. As HTS technologies are adopted into other related disciplines compound potency and selectivity are no longer the only parameters to be optimized during hit-finding. With this broader acceptance of key technologies, harmonization and standardization of data across disciplines are crucial to facilitate analysis and mining of the data. Important information such as compound purity and its associated physico-chemical properties such as solubility can be derived very quickly on relatively large numbers of compounds and thus help prioritize compounds for progression based on overall suitability, not just potency (Fligge and Schuler, 2006). These quality criteria, and quality assessment at all key points in the discovery process, are crucial. Late-stage attrition of drug candidates, particularly in development and beyond, is extremely expensive and such failures must be kept to a minimum. This is typically done by an extensive assessment of chemical integrity, synthetic accessibility, functional properties, structure–activity relationship (SAR) and biophysicochemical properties, and related absorption, distribution, metabolism and excretion (ADME) characteristics, as discussed further in Chapters 9 and 10.
Lead discovery and high-throughput screening
A lead compound is generally defined as a new chemical entity that could potentially be developed into a new drug by optimizing its beneficial effects and minimizing its side effects (see Chapter 9 for a more detailed discussion of the criteria). HTS is currently the main approach for the identification of lead compounds, i.e. large numbers of compounds (the ‘compound library’) are usually tested in a random approach for their biological activity against a disease-relevant target. However, there are other techniques in place for lead discovery that are complementary to HTS.
Besides the conventional literature search (identification of compounds already described for the desired activity), structure-based virtual screening is a frequently applied technique (Ghosh et al., 2006; Waszkowycz, 2008). Molecular recognition events are simulated by computational techniques based on knowledge of the molecular target, thereby allowing very large ‘virtual’ compound libraries (greater than 4 million compounds) to be screened in silico and, by applying this information, pharmacophore models can be developed. These allow the identification of potential leads in silico, without experimental screening and the subsequent construction of smaller sets of compounds (‘focused libraries’) for testing against a specific target or family of targets (Stahura et al., 2002; Muegge and Oloff, 2006). Similarly, X-ray analysis of the target can be applied to guide the de novo synthesis and design of bioactive molecules. In the absence of computational models, very low-molecular-weight compounds (typically 150–300 Da, so-called fragments), may be screened using biophysical methods to detect low-affinity interactions. The use of protein crystallography and X-ray diffraction techniques allows elucidation of the binding mode of these fragments and these can be used as a starting point for developing higher affinity leads by assemblies of the functional components of the fragments (Rees et al., 2004; Hartshorn et al., 2005; Congreve et al., 2008).
Typically, in HTS, large compound libraries are screened (‘primary’ screen) and numerous bioactive compounds (‘primary hits’ or ‘positives’) are identified. These compounds are taken through successive rounds of further screening (’secondary’ screens) to confirm their activity, potency and where possible gain an early measure of specificity for the target of interest. A typical HTS activity cascade is shown in Figure 8.2 resulting in the identification of hits, usually with multiple members of a similar chemical core or chemical series. These hits then enter into the ‘hit-to-lead’ process during which medicinal chemistry teams synthesize specific compounds or small arrays of compounds for testing to develop an understanding of the structure–activity relationship (SAR) of the underlying chemical series. The result of the hit-to-lead phase is a group of compounds (the lead series) which has appropriate drug-like properties such as specificity, pharmacokinetics or bioavailability. These properties can then be further improved by medicinal chemistry in a ‘lead optimization’ process (Figure 8.3). Often the HTS group will provide support for these hit-to-lead and lead optimization stages through ongoing provision of reagents, provision of assay expertise or execution of the assays themselves.
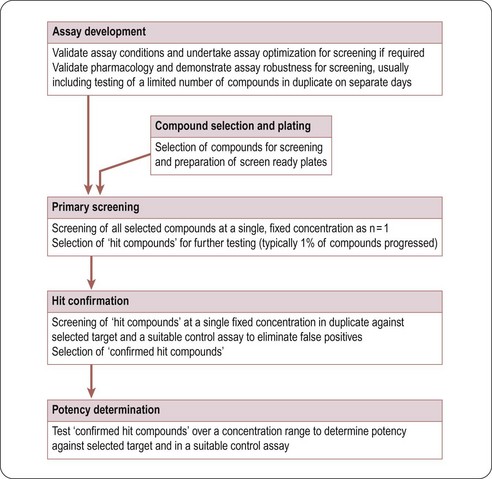
Assay development and validation
The target validation process (see Chapters 6 and 7) establishes the relevance of a target in a certain disease pathway. In the next step an assay has to be developed, allowing the quantification of the interaction of molecules with the chosen target. This interaction can be inhibition, stimulation, or simply binding. There are numerous different assay technologies available, and the choice for a specific assay type will always be determined by factors such as type of target, the required sensitivity, robustness, ease of automation and cost. Assays can be carried out in different formats based on 96-, 384-, or 1536-well microtitre plates. The format to be applied depends on various parameters, e.g. readout, desired throughput, or existing hardware in liquid handling and signal detection with 384- (either standard volume or low volume) and 1536-well formats being the most commonly applied. In all cases the homogeneous type of assay is preferred, as it is quicker, easier to handle and cost-effective, allowing ‘mix and measure’ operation without any need for further separation steps.
Next to scientific criteria, cost is a key factor in assay development. The choice of format has a significant effect on the total cost per data point: the use of 384-well low-volume microtitre plates instead of a 96-well plate format results in a significant reduction of the reaction volume (see Table 8.1). This reduction correlates directly with reagent costs per well. The size of a typical screening library is between 500 000 and 1 million compounds. Detection reagent costs per well can easily vary between US$0.05 and more than U$0.5 per data point, depending on the type and format of the assay. Therefore, screening an assay with a 500 000 compound library may cost either US$25 000 or US$250 000, depending on the selected assay design – a significant difference! It should also be borne in mind that these costs are representative for reagents only and the cost of consumables (assay plates and disposable liquid handling tips) may be an additional consideration. Whilst the consumables costs are higher for the higher density formats, the saving in reagent costs and increased throughput associated with miniaturization usually result in assays being run in the highest density format the HTS department has available.
Table 8.1 Reaction volumes in microtitre plates
| Plate format | Typical assay volume |
|---|---|
| 96 | 100–200 µL |
| 384 | 25–50 µL |
| 384 low volume | 5–20 µL |
| 1536 | 2–10 µL |
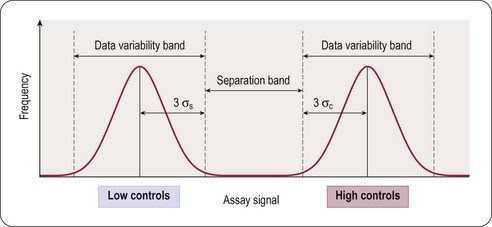
The principal goal of developing HTS assays is the fast and reliable identification of active compounds (‘positives’ or ‘hits’) from chemical libraries. Most HTS programmes test compounds at only one concentration. In most instances this approximates to a final test concentration in the assay of 10 micromolar. This may be adjusted depending on the nature of the target but in all cases must be within the bounds of the solvent tolerance of the assay determined earlier in the development process. In order to identify hits with confidence, only small variations in signal measurements can be tolerated. The statistical parameters used to determine the suitability of assays for HTS are the calculation of standard deviations, the coefficient of variation (CV), signal-to-noise (S/N) ratio or signal-to-background (S/B) ratio. The inherent problem with using these last two is that neither takes into account the dynamic range of the signal (i.e. the difference between the background (low control) and the maximum (high control) signal), or the variability in the sample and reference control measurements. A more reliable assessment of assay quality is achieved by the Z’-factor equation (Zhang et al., 1999):

This equation takes into account that the quality of an assay is reflected in the variability of the high and low controls, and the separation band between them (Figure 8.3). Z’-factors are obtained by measuring plates containing 50% low controls (in the protease example: assay plus reference inhibitor, minimum signal to be measured) and 50% high controls (assay without inhibitor; maximum signal to be measured). In addition, inter- and intra-plate coefficients of variation (CV) are determined to check for systematic sources of variation. All measurements are normally made in triplicate. Once an assay has passed these quality criteria it can be transferred to the robotic screening laboratory. A reduced number of control wells can be employed to monitor Z’-values when the assay is progressed to HTS mode, usually 16 high- and 16 low-controls on a 384-well plate, with the removal of no more than two outlying controls to achieve an acceptable Z’-value. The parameter can be further modified to calculate the Z-value, whereby the average signal and standard deviation of test compound wells are compared to the high-control wells (Zhang et al., 1999). Due to the variability that will be present in the compound wells, and assuming a low number of active compounds, the Z-value is usually lower than the Z’-value.
Whilst there are been several alternatives of Zhang’s proposal for assessing assay robustness, such as power analysis (Sui and Wu, 2007), the simplicity of the equation still make the Z’-value the primary assessment of assay suitability for HTS.
The Assay Guidance Website hosted by the National Institutes of Health Center for Translational Therapeutics (NCTT) (http://assay.nih.gov/assay/index.php/Table_of_Contents) provides comprehensive guidance of factors to consider for a wide range of assay formats.
Biochemical and cell-based assays
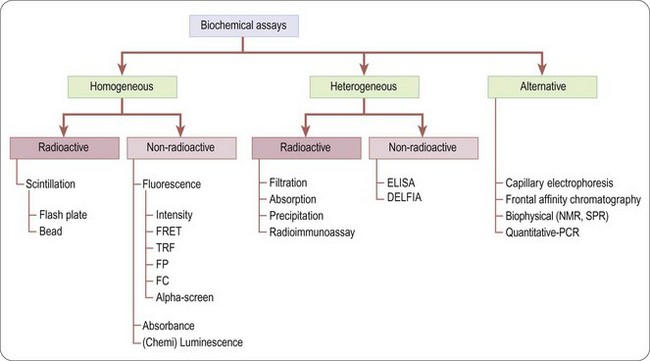
There is a wide range of assays formats that can be deployed in the drug discovery arena (Hemmilä and Hurskainen, 2002), although they broadly fall into two categories: biochemical and cell-based.
Biochemical assays (Figure 8.4) involve the use of cell-free in-vitro systems to model the biochemistry of a subset of cellular processes. The assay systems vary from simple interactions, such as enzyme/substrate reactions, receptor binding or protein–protein interactions, to more complex models such as in-vitro transcription systems. In contrast to cell-based assays, biochemical assays give direct information regarding the nature of the molecular interaction (e.g. kinetic data) and tend to have increased solvent tolerance compared to cellular assays, thereby permitting the use of higher compound screening concentration if required. However, biochemical assays lack the cellular context, and are insensitive to properties such as membrane permeability, which determine the effects of compounds on intact cells.
Unlike biochemical assays, cell-based assays (Figure 8.5) mimic more closely the in-vivo situation and can be adapted for targets that are unsuitable for screening in biochemical assays, such as those involving signal transduction pathways, membrane transport, cell division, cytotoxicity or antibacterial actions. Parameters measured in cell-based assays range from growth, transcriptional activity, changes in cell metabolism or morphology, to changes in the level of an intracellular messenger such as cAMP, intracellular calcium concentration and changes in membrane potential for ion channels (Moore and Rees, 2001). Importantly, cell-based assays are able to distinguish between receptor antagonists, agonists, inverse agonists and allosteric modulators which cannot be done by measuring binding affinity in a biochemical assay.
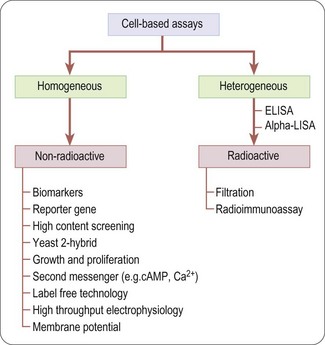
Cell-based assays frequently lead to higher hit rates, because of non-specific and ‘off-target’ effects of test compounds that affect the readout. Primary hits therefore need to be assessed by means of secondary assays such as non- or control-transfected cells in order to determine the mechanism of the effect (Moore and Rees, 2001).
Although cell-based assays are generally more time-consuming than cell-free assays to set up and run in high-throughput mode, there are many situations in which they are needed. For example, assays involving G-protein coupled receptors (GPCRs), membrane transporters and ion channels generally require intact cells if the functionality of the test compound is to be understood, or at least membranes prepared from intact cells for determining compound binding. In other cases, the production of biochemical targets such as enzymes in sufficient quantities for screening may be difficult or costly compared to cell-based assays directed at the same targets. The main pros and cons of cell-based assays are summarized in Table 8.2.
Table 8.2 Advantages and disadvantages of cell-based assays
| Advantages | Disadvantages |
|---|---|
| Cytotoxic compounds can be detected and eliminated at the outset | Require high-capacity cell culture facilities and more challenging to fully automate |
| In receptor studies, agonists can be distinguished from antagonists | Often require specially engineered cell lines and/or careful selection of control cells |
| Detection of allosteric modulators | Reagent provision and control of variability of reagent batches |
| Binding and different functional readouts can be used in parallel – high information content | Cells liable to become detached from support |
| Phenotypic readouts are enabling when the molecular target is unknown (e.g. to detect compounds that affect cell division, growth, differentiation or metabolism) | High rate of false positives due to non-specific effects of test compounds on cell function |
| More disease relevant than biochemical asays | Assay variability can make assays more difficult to miniaturize |
| No requirement for protein production/scale up | Assay conditions (e.g. use of solvents, pH) limited by cell viability |
Assay readout and detection
Ligand binding assays
The majority of homogenous radioactive assay types are based on the scintillation proximity principle. This relies on the excitation of a scintillant incorporated in a matrix, in the form of either microbeads (’SPA’) or microplates (Flashplates™, Perkin Elmer Life and Analytical Sciences) (Sittampalam et al., 1997), to the surface of which the target molecule is also attached (Figure 8.6). Binding of the radioligand to the target brings it into close proximity to the scintillant, resulting in light emission, which can be quantified. Free radioactive ligand is too distant from the scintillant and no excitation takes place. Isotopes such as 3H or 125I are typically used, as they produce low-energy particles that are absorbed over short distances (Cook, 1996). Test compounds that bind to the target compete with the radioligand, and thus reduce the signal.
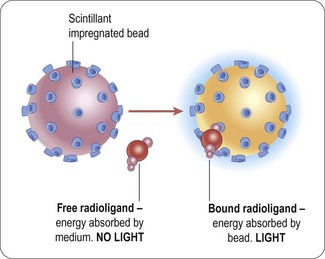
Fig. 8.6 Principle of scintillation proximity assays.
Reproduced with kind permission of GE Healthcare.
With bead technology (Figure 8.8A), polymer beads of ~5 µm diameter are coated with antibodies, streptavidin, receptors or enzymes to which the radioligand can bind (Bosworth and Towers, 1989; Beveridge et al., 2000). Ninety-six- or 384-well plates can be used. The emission wavelength of the scintillant is in the range of 420 nm and is subject to limitations in the sensitivity due to both colour quench by yellow test compounds, and the variable efficiency of scintillation counting, due to sedimentation of the beads. The homogeneous platforms are also still subject to limitations in throughput associated with the detection technology via multiple photomultiplier tube-based detection instruments, with a 384-well plate taking in the order of 15 minutes to read.
The drive for increased throughput for radioactive assays led to development of scinitillants, containing europium yttrium oxide or europium polystyrene, contained in beads or multiwell plates with an emission wavlength shifted towards the red end of the visible light spectrum (~560 nm) and suited to detection on charge-coupled device (CCD) cameras (Ramm, 1999). The two most widely adopted instruments in this area are LEADseeker™ (GE Healthcare) and Viewlux™ (Perkin Elmer), using quantitative imaging to scan the whole plate, resulting in a higher throughput and increased sensitivity. Imaging instruments provide a read time typically in the order of a few minutes or less for the whole plate irrespective of density, representing a significant improvement in throughput, along with increased sensitivity. The problem of compound colour quench effect remains, although blue compounds now provide false hits rather than yellow. As CCD detection is independent of plate density, the use of imaging based radioactive assays has been adopted widely in HTS and adapted to 1536-well format and higher (see Bays et al., 2009, for example).
In the microplate form of scintillation proximity assays the target protein (e.g. an antibody or receptor) is coated on to the floor of a plate well to which the radioligand and test compounds are added. The bound radioligand causes a microplate surface scintillation effect (Brown et al., 1997). FlashPlate™ has been used in the investigation of protein–protein (e.g. radioimmunoassay) and receptor–ligand (i.e. radioreceptor assay) interactions (Birzin and Rohrer, 2002), and in enzymatic (e.g. kinase) assays (Braunwaler et al., 1996).
Due to the level of sensitivity provided by radioactive assays they are still widely adopted within the HTS setting. However, environmental, safety and local legislative considerations have led to the necessary development of alternative formats, in particular those utilizing fluorescent-ligands (Lee et al., 2008; Leopoldo et al., 2009). Through careful placement of a suitable fluorophore in the ligand via a suitable linker, the advantages of radioligand binding assays in terms of sensitivity can be realized without the obvious drawbacks associated with the use of radioisotopes. The use of fluorescence-based technologies is discussed in more detail in the following section.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


