Fig. 2.1
Representative differential scanning fluorometry spectra obtained with a therapeutic protein on 96-well plate filled with different formulations for each well. Fluorescence intensity in relative fluorescence units (RFU) is shown as a function of temperature. Protein concentration is 1 g/L. The sample volume is 30 μL. Data were obtained using the Bio-Rad CFX96 RT-PCR plate reader
2.2.8 Light Scattering
Aggregation is one of the major problems in protein pharmaceutical development. The presence of aggregated protein can compromise the purity, safety, and efficacy of a drug product. Several separation and detection techniques are used to monitor protein aggregation. Light scattering methods have the advantage of high sensitivity due to the size of scatterers, which makes it possible to detect small amounts of large protein aggregates in pharmaceutically relevant samples. There are two basic types of such methods used in protein therapeutics development: static light scattering (SLS) and dynamic light scattering (DLS). SLS can be applied to determine the protein’s molecular mass and the mean square radius of gyration. SLS is often used with separation methods such as SEC or field-flow fractionation (FFF) for the purpose of obtaining a more accurate estimate of the size of the components separated by these techniques (Tarazona and Saiz 2003; McEvoy et al. 2011). Such online scattering method is commonly known as multi-angle light scattering (MALS). The throughput of SLS analysis used in this fashion often depends on the throughput of the separation technique. Though significant advancements have been achieved in numerous types of separation technologies, a variety of physical parameters, such as high pressure and short equilibration times, can be problematic for the coupling of light scattering detectors. Light scattering can also be coupled to plate readers. Simple monitoring of the scattered light at fixed wavelengths and angles can provide sensitive detection of aggregates formation. The Stargazer-384TM system (Harbinger Biotechnology and Engineering Corporation, Toronto, Canada), a 384-well microplate reader, was used in a study to monitor colloidal stability of mAbs at elevated temperatures (Goldberg et al. 2010). As previously discussed, the high-throughput Avacta Optim® 1000 (Pall Corporation, Port Washington, NY) is available for light scattering as well as fluorescence measurements with a volume as low as 1 μL. In these instruments the light scattering signal is used to monitor aggregation by detecting the increased intensity, similar to turbidity measurements but with higher sensitivity. It should be noted, however, that if the aggregates formed are less dense than the monomeric protein, decreases in scattering can also be seen.
Dynamic light scattering is based on the measurement of the fluctuations in intensity of scattered light. An autocorrelation function, derived from fluctuation analysis, can reveal the distribution of the hydrodynamic radii of protein molecules present in solution (Schmidt 2010). Separation is not necessary, but resolution of species depends on their size difference and concentration. Because of the exponential form of the autocorrelation function, no more than two to four components can be comfortably resolved in the same solution. DLS plate readers have become very popular in the biopharmaceutical industry for protein and vaccine characterization (Vincentelli et al. 2004). Multiwell plate formats, small volumes, and automated procedures for measurement and data analysis make the DLS method high throughput and easy to apply. Antibody self-association has been studied with the help of gold nanoparticles and their characterization by dynamic and static light scattering. Nanoparticle–antibody conjugates displayed complex aggregation behavior dependent on pH and ionic strength of the solution. Use of a DLS plate reader was a significant part of the high-throughput analytical development (Sule et al. 2011).
In biopharmaceutical drug development DLS has been used not only for direct detection and characterization of aggregation but also for the study of large colloidal structures. Certain large colloid-like aggregates have been shown to inhibit enzymes leading to false-positive HTS leads. These so-called promiscuous inhibitors were detected and screened by DLS using a plate reader (Feng et al. 2005). Results from such high-throughput assays for promiscuous inhibitory aggregates have been used to develop new computational models of this phenomenon. A method for quantitative characterization of macromolecular interactions using DLS has been introduced in a temperature-controlled plate reader format (Hanlon et al. 2010). This technique enabled determination of equilibrium dissociation constants and thermodynamic parameters. The low volume of plate-based DLS reduced the sample amount to a few microliters per experiment, with detection limits in the femtomolar range.
Biopharmaceutical products are often formulated at high concentrations to maximize delivery dosage and efficiency, and solutions of some proteins become very viscous at high concentrations (Yadav et al. 2010), creating significant problems for processes like purification, filtration, and injection through syringes. Standard methods for viscosity measurements have low throughput and require large quantities of protein. Thus, there is increasing demand for higher-throughput viscosity screening. A DLS assay based on measurement of the diffusion coefficient of beads added directly to the protein solution is high throughput and run in a multiwell plate format (He et al. 2010a). As shown in Fig. 2.2 the Stokes–Einstein equation can be used to calculate the viscosity of a protein solution using the known radius of the added beads and the measured diffusion coefficient. Furthermore, DLS measurements of diffusion coefficients as a function of protein concentration can be used to derive the interaction parameter, k D , which has been shown to correlate with protein properties such as viscosity and particulation propensity (Yadav et al. 2010; He et al. 2011). It is widely accepted that the second virial coefficient, B22, obtained by SLS measurements contains information on protein–protein interaction (Printz et al. 2012). The k D parameter derived from DLS measurements offers a simple way to compare samples under similar conditions (discussed in Chap. 3).
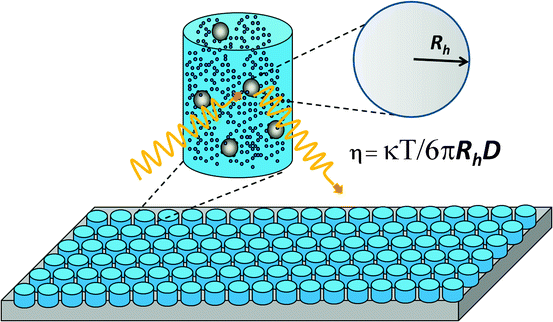
Fig. 2.2
High-throughput method for viscosity measurements based on dynamic light scattering determination of the diffusion coefficient of polystyrene beads externally added into a protein solution
2.2.9 Design of Experiment and Data Analysis
All high-throughput methodologies mentioned above share a common ability to generate a large amount of biophysical information on therapeutic proteins. This enables more complex experimental designs at various stages of pharmaceutical development. The Quality by Design (QbD) concept has gained popularity in recent years among the biopharmaceutical industry and regulatory agencies (Rathore and Winkle 2009; Rathore and Devine 2008). Design of product quality is built on comprehensive understanding of a well-defined process and product space where the protein therapeutic is in its most desired form. The principle of design of experiment (DOE) is often applied to systematically evaluate the protein of interest under a variety of conditions, which are often selected based on the types of stresses that a protein therapeutic is subjected to during manufacturing, storage, and administration. The degree of these stresses often exceeds reality, and the experimental results can be used to predict the protein behavior when failures occur during the product life cycle. The combination of high-throughput measurement and DOE can also help enhance the statistical power when interpreting the results. More importantly, the implementation of high-throughput methodology offers opportunities to simultaneously assess a large number of samples. This is particularly valuable when employing methods that are only used to qualitatively rank order protein samples.
Application of high-throughput techniques results in large data sets, often requiring mathematical tools for rigorous analysis. Statistics helps to establish correlations among measured properties of a molecule. Commonly used statistical analyses that are frequently applied to the development of protein therapeutics include Gaussian modeling, analyses of variance (ANOVA), and the t-test. In addition, biophysical characterization often involves spectroscopic methods such as CD, FTIR, and fluorescence, and the results usually include measured values as a function of wavelength of the optic source. Such data can be analyzed by methods of chemometrics including the singular value decomposition technique which has been used to determine the maximum changes in protein properties caused by particular factors like pH or ionic strength. This methodology has been applied to the interpretation of CD and FTIR spectra obtained during the production of antibodies (Greenfield 2006; Sellick et al. 2010). Another useful mathematical tool is polynomial-based data fitting. This approach involves fitting an arbitrary polynomial model to a limited data set and then using the same model to predict protein behavior outside of the tested range. For example, discrete data at pH 5, 5.5, and 6 can be used to generate a polynomial model that best fit the experimental results. The continuous mathematical model can then be used to predict results at pH 5.8, and even at pH values outside of 5–6, if the assumption holds. The polynomial method is especially effective when the source data set is large. Even more predictive information can be obtained while considering multiple variables simultaneously (Sall et al. 2007).
2.3 Empirical Phase Diagrams as Tools to Interpret Results from High-Throughput Biophysical Approaches
2.3.1 Combination of Biophysical Techniques and Data Analysis
As discussed above and in other chapters in this text, high-throughput screening (HTS) is usually performed with only one or two low-resolution techniques of the type previously considered. The selection of the particular technique is typically based on what is known about degradation pathways of the target, convenience, and availability of appropriate instrumentation as well as speed. It has recently become possible to combine the results from multiple techniques with the goal of providing a more comprehensive picture of a protein’s structure and its response to various environmental perturbations. This can be used to select optimal methods for HTS as well as for various forms of comparative analysis. A number of methods are available for this purpose. We will consider here only the one that has been most thoroughly described in the literature [reviewed in Maddux et al. (2011)], but other approaches such as Chernoff faces and star charts (Yau 2011) in which information is encoded in facial features or abstract geometric shapes are under consideration. The former method is known as the EPD. The word empirical is inserted in front of the phrase “phase diagram” to differentiate it from the well-known thermodynamic or equilibrium phase diagram since equilibrium conditions are not implied in the former.
The basic idea behind the EPD is to represent the protein (application has also been made to peptides, nucleic acids, virus-like particles, viruses, and bacteria cells) as a vector in which the components of the vector are experimental values obtained from the various methods employed as a function of solution variables. Preparation of EPDs generally involves buffer subtraction of the data, peak selection for data analysis (entire spectra can also be used), averaging of multiple data acquisitions, normalization, input matrix synthesis, singular value decomposition, and finally color mapping of the most significant data using an RGB color scheme. A detailed description of the method including the mathematics involved is presented in Maddux et al. (2011). In general, far-UV circular dichroism (CD) is used to monitor secondary structure although FTIR and Raman spectroscopies can also be employed. Tertiary structure is most commonly analyzed with intrinsic fluorescence, near-UV CD, or high-resolution derivative absorption spectroscopy. Dye binding using compounds such as 8-anilino naphthalene sulfonic acid (ANS) is frequently used to probe the exposure of apolar regions in the protein. Dissociation and association (including aggregation) are typically probed with static and/or DLS (although see below). Overall thermal stability is often studied with differential scanning calorimetry. The most common independent variables (i.e., forms of stress that have previously been employed) are temperature and pH although a wide variety of other variables have been used as described below. Perhaps the major limitation of the EPD approach has been the time and instrumentation necessary to prepare an EPD. This has recently changed with the advent of equipment capable of performing multiple different types of measurements simultaneously. Originally an EPD typically required a fluorometer, CD spectropolarimeter, light scattering system, and perhaps a DSC or FTIR spectrometer. Several newly developed instruments now at least partially overcome this limitation. For example, recent improvements in CD instruments now permit both near- and far-UV spectra, near-UV absorption spectra, fluorescence, and SLS (turbidity or scattering at the fluorescence emission wavelength) to all be acquired simultaneously in a four-position sample chamber under variable temperature conditions (Hu et al. 2011). This permits EPDs to be generated in less than a day. A similar “protein machine” with a six-position sample chamber has also been recently described (Maddux et al. 2012). Perhaps the simplest version of a system with rapid EPD generation capability is a UV absorption spectrometer, typically of the diode array variety, to permit sufficient resolution of derivative peaks (Kueltzo et al. 2003). At high resolution (usually second), the derivative spectrum of a protein will usually manifest distinct peaks for phenylalanine, tyrosine, and tryptophan (if present). Since the residues are usually buried, Tyr is often interfacial, and Trp present in highly variable environments, temperature, and pH-induced peaks shifts can frequently provide a fairly detailed picture of a protein’s structural response to various perturbations. Such data can easily be represented in the form of an EPD (Kueltzo et al. 2003). Similarly, fluorescence microtiter plate-based fluorometers have been developed which employ multiple fluorescence and light scattering measurements as a function of temperature, as also described in the fluorescence section. The latter is of especially high throughput, permitting the generation of many EPDs in a single day. EPDs for four model proteins obtained from multiple instruments, two CD-based spectrometers, and a high-throughput fluorometer are shown in Fig. 2.3, where it can be seen that all produce similar EPDs although small differences are apparent due to the various types of measurements used to construct each EPD.
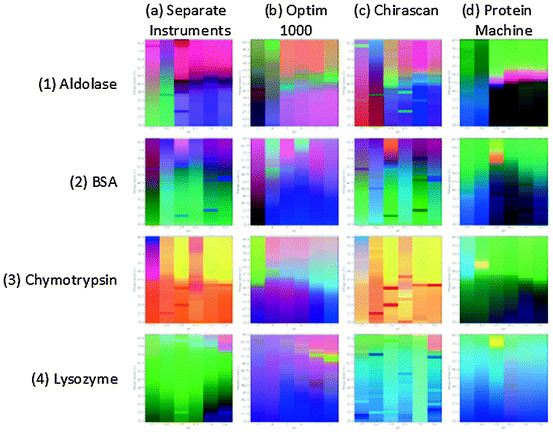
Fig. 2.3
Empirical phase diagrams (EPDs) of four model proteins (1) aldolase, (2) BSA, (3) chymotrypsin, and (4) lysozyme constructed using data collected from various instruments: (a) intrinsic fluorescence (FL) and static light scattering (SLS) data from a Photon Technology International (PTI) fluorometer, and circular dichroism (CD) from an Applied Photophysics Chirascan, (b) FL and SLS by an Avacta Optim 1000 microtiter plate fluorometer, (c) FL and CD Applied Photophysics Chirascan, and (d) FL, SLS, CD, and UV absorbance by an Olis Protein Machine
2.3.2 High-Throughput Characterization and Preformulation Development
The EPD method provides a comprehensive overview of how a protein responds to environmental alteration in the form of a colored diagram in which regions of different color correspond to different structural states of the target molecules. By reference to the original data, native partially folded and molten globule, extensively unfolded, dissociated, oligomerized, and various aggregated states can all be identified. This provides the scientist with clues to trouble spots in a protein and a basis with which to select assays with which to screen for potential stabilizers. For example, if aggregation or a particular structural change occurs under moderate temperature and/or pH conditions, one can select a less stable condition and one or more techniques sensitive to selected degradation events for screening purposes. Typically, a supplemental GRAS (generally regarded as safe) library containing a selection of buffers, sugars, sugar alcohols, amino acids, polymers, detergents, and osmolytes is used. In the initial screen relatively high concentrations of compounds are used with their concentration dependence and use in combination later optimized. It is usually wise to employ at least two methods: one sensitive to aggregation (e.g., light scattering) and one to structural change (e.g., fluorescence, CD) for this purpose. DSC is also commonly employed especially due to the recent availability of highly sensitive high-throughput instruments. It is also possible to prepare EPDs in the presence of selected stabilizers to permit a more detailed analysis/comparison of their effects on a protein. The information thus obtained by a temperature/pH EPD thus provides a basis for buffer and excipient selection at an early stage of pharmaceutical development. Although not yet published, a new version of the EPD has been developed in which the colors have actual physical measuring in contrast to the arbitrary assignment of color in the original EPD.
2.3.3 Additional Application of High-Throughput Methods and the EPD
High-throughput methods and EPDs can also be applied to a wide variety of different situations, some of which will be briefly described here. Two commonly encountered forms of stress in the protein therapeutic area are freeze/thaw and shear. It is often necessary to freeze and then thaw both during development and manufacturing situations. An EPD can be created using the number of freeze/thaw cycles under defined conditions as an independent variable accompanied by temperature and pH stress. All three variables (temperature, pH, freeze/thaw cycles) can be combined into a three-dimensional representation in which the EPD is presented as a colored surface. Shear stress is also often encountered in the development, manufacture (especially filling), and shipping of protein pharmaceuticals. To explore this potential degrading stress, the intensity of the shear can be varied by a mechanical process such as stirring, shaking, or some other forms of agitation, and this is used as a variable in EPD production.
Protein concentration is another important variable that has assumed increasing importance with the use of high-concentration formulations. This variable can be typically evaluated over the range of 0.05–300 g/L depending on the solubility of the protein and the methods employed in the analysis. Proteins usually alter their structure to little or no extent as a function of protein concentration, but aggregation and surface adsorption are both highly concentration dependent. Thus, aggregation-sensitive techniques such as light scattering are often of special importance in protein concentration-dependent studies. Another common variable of particular importance is ionic strength. In the Debye–Huckel charge shielding regime (0–0.15 ionic strength), a number of intermediate concentrations should be evaluated to probe electrostatic effects. At higher salt concentrations both preferential hydration and binding effects usually dominate with salt concentration into the molar range appropriately examined.
It is also possible to create EPDs based on phenomena such as aggregation. A variety of different types of aggregates have been identified in protein solutions based on their relative size and the nature of a protein’s conformation (altered or native) within the aggregate. A number of methods are available that are sensitive to these features (see Chap. 9), permitting an aggregation-based EPD to be created. Again, using variables like temperature, pH, ionic strength, freeze/thaw, and shear, soluble protein aggregates can be detected by methods such as size-exclusion HPLC, sedimentation velocity analytical ultracentrifugation, FFF, and DLS. Complimentary structural data can be obtained by the methods described above with FTIR and Raman spectroscopy especially useful because of the particulate nature of such samples. Larger (i.e., submicron) aggregates can be characterized by DLS including single-particle microscope-based approaches (nanoparticle tracking analysis) and classic microscopy-based techniques (atomic force microscopy, scanning electron microscopy, and transmission electron microscopy) although they are difficult to quantitate and new methods such as quartz crystal microbalances and nanomechanical resonators are seeing increasing use. Subvisible and visible particles can be analyzed by methods such as coulter counting, light obscuration, micro-flow digital imaging, and various visual procedures. Using parameters such as size, composition, structure, and particle number as dependent variables, an EPD can be generated that provides a comprehensive picture of the nature of protein aggregates that form under a wide variety of stress conditions.
Although not yet described in the scientific literature, chemical degradation can also be analyzed in various high-throughput modes and be summarized in EPDs. The application of EPDs described above has all been to various physical processes in which covalent bonds are not broken. Of equal importance to protein degradation, however, are chemical changes such as oxidation and deamidation events. Chemical changes are usually quantitatively determined by peptide mapping combined with mass spectrometry (MS) in the form of HPLC-MS experiments which permit both the amount and location of a residue modification within a protein’s amino acid sequence to be determined. To increase throughput, once the nature of any changes has been identified, HPLC-MS analysis may be replaced by methods such as RP-HPLC and capillary electrophoresis or isoelectric focusing. A convenient way to present and analyze such data is in the form of rate constants for the individual, for example, deamidation and oxidation events. This of course requires time-dependent measurements. The rate constants can be used as the dependent variables in an EPD since they are typically determined as function-independent variables such as pH and temperature. Of special interest is the comparative use of physical and chemical EPDs which permits an exploration of the relationship between structural changes and chemical events (and vice versa).
As a final example, EPDs can be used in a strictly comparative mode. Structural comparisons between both similar and assumed identical proteins are often a very important element of pharmaceutical analysis. For example, when manufacturing changes are made, during the development of biosimilars or when investigating mutant proteins, a detailed comparison of the various species is a critical part of the analysis. A direct comparison of EPDs of the target molecules provides a convenient and sensitive way to see if structural identity has been obtained. As an example, second-generation functional mutants of fibroblast growth factor one (FGF-1) have been compared using EPDs and used to select molecules that are not dependent on heparin for their activity (Alsenaidy et al. 2012). Such EPD-dependent comparisons have even been performed with different rotavirus serotypes despite their individual complexity (Esfandiary et al. 2010). Various mathematical (difference) methods exist to facilitate such comparisons.
2.4 Advantages and Challenges of Implementing High-Throughput Technology in Therapeutic Protein Development: An Industry Perspective
High-throughput technologies can be applied across all aspects of biopharmaceutical discovery and development from the early stages of candidate screening and selection to the later stages of formulation development. The benefits of a successful high-throughput screening strategy in this environment are many. The most obvious one is speed. Faster assays mean that sufficient data to drive a decision can be collected within a shorter timeframe leading to more rapid decisions and ultimately an accelerated development timeline. In the pharmaceutical industry where product development is a protracted process, any acceleration of the timeline can mean getting a promising drug candidate into clinical testing sooner and ultimately to market faster, potentially providing a competitive advantage and leading to an earlier revenue stream. A second advantage is that high-throughput assays typically require smaller volumes and fewer samples than standard assay formats. This sample sparing feature is especially important early in development in which the purification process is an early stage of development and consequently the quantities of the candidates being tested may be in short supply. Another advantage is that high-throughput procedures permit many more drug candidates to be tested than would be possible using a standard approach. In the early stages of discovery research where many candidates are being evaluated based on screening assays, a good high-throughput screen for binding permits many more candidates to be tested and allows this to be accomplished within a shorter period of time. Finally, high-throughput assays permit additional molecular features to be evaluated. A good example of this advantage is found in formulation development where the influence of a variety of solution conditions and potential excipients will need to be tested. The solution pH, buffer salts used, excipients, surfactants, and the presence or absence of salt all need to be evaluated, and this can be accomplished much more easily using high-throughput approaches.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


