CHAPTER 31 Hemostasis
Principles of investigation
Introduction
Hemostatic tests retain unique features and problems in interpretation. Even coagulation screening tests (the only commonly requested tests that require explicit coreporting of control experiments) are complex bioassays in miniature. An abnormal value can have diametrically opposed meanings for patient care depending on the clinical context. Expressing clinical pretest probability in an intelligible way and using test results to modify this probability is the best way of avoiding potential confusion and error.1
The application of meta-analysis of randomized studies (‘evidence-based medicine’) to diagnostic laboratory testing has been limited,2 and hemostatic testing is no exception. It is therefore not possible yet to claim evidence-based validation, in its strict sense, for many of the principles discussed below. However, the writings of many expert clinician–scientists over the years are the best guide we have to these principles, and should certainly form a starting-point for further analyses.
Physiology of hemostasis applied to diagnosis
To assist this diagnostic thinking, it helps to keep in mind a simplified map of the hemostatic system, whatever knowledge of its complexity one possesses (or not, as the case may be). These simple maps are caricatures: readers are referred to fuller versions3,4 and to other chapters in this volume.
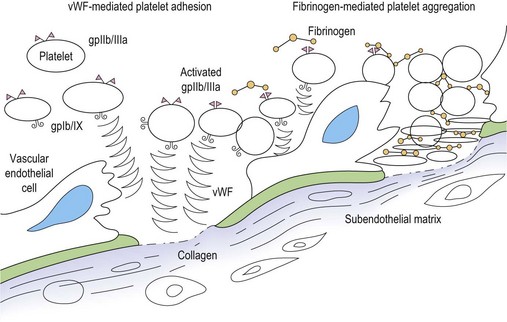
Primary hemostasis: formation of the platelet plug5 (Fig. 31.1)
Secondary hemostasis: generation of fibrin clot by the coagulation pathway (Fig. 31.2)
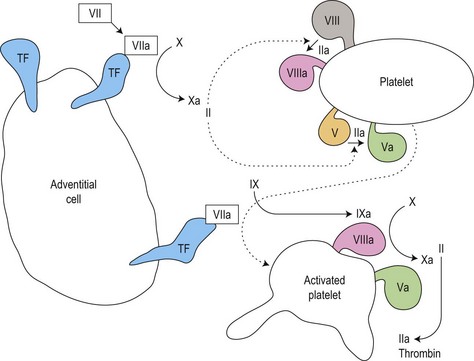
Fig. 31.2 Thrombin generation in vivo: coagulation factor interactions mediated by cell surfaces (schematic).
Clot regulation and removal: the protein C and fibrinolytic pathways
Two further systems regulate and eventually remove the clot (in the context of tissue repair and neoangiogenesis) (also see Chapter 28):
Clot regulation: the protein C system6 and antithrombin
Thrombin activity is restricted to the platelet surface and the fibrin matrix of the clot by a conformation-dependent inhibitor, antithrombin (AT)7 that inactivates fluid-phase thrombin. To work efficiently, AT must bind to heparin-like proteoglycans on healthy endothelial cells.
Fibrinolysis: the plasmin system8
Clots contain the seeds of their own destruction in the form of a clot-bound protein, plasminogen. This is cleaved by tissue plasminogen activator (tPA) secreted by healthy vascular endothelial cells, or urokinase on the surface of macrophages, to the fibrinolytic enzyme plasmin. Plasmin cleaves fibrin into fibrin degradation products, notably the D-dimer fragment specific to cleavage of cross-linked fibrin.9 This removes the clot, and activates matrix metalloproteinases that initiate remodeling of vessels (angiogenesis).
The clinical approach to the patient with a possible hemostatic disorder
Experts writing about the investigation of possible bleeding disorders unanimously stress the importance of a carefully taken history.10–12 They also recommend specific questions, answers to which alter the pretest probability of a bleeding disorder. The discussion below draws on this consensus. Similarly, key findings on clinical examination may aid the diagnostic process, although they are less frequent than narrative clues.
It must be conceded that these narrative and clinical signs have not been formally tested, either singly or in clusters, for their relative value in predicting the presence of hemostatic disorders. Such testing has refined and simplified the use of clinical clues in other contexts,13 and may be of future benefit in hemostasis. Until such clarification becomes available, the shared insight of experienced clinicians is our best guide.
Key questions
Surgical challenges
Dental surgery
Surgical trauma to the incompressible tooth socket sitting in the fibrinolytic milieu of the oral cavity is a stiff challenge to the hemostatic system. Useful questions about the effect of extractions focus on the duration of bleeding and the actions compelled by it. Compare the two accounts in Box 31.1. The second account gives a much clearer indication of excessive blood loss. Most people (in the UK, at least) are disinclined to make an early return to the dentist without a pressing reason.
Bruising
A patient complaining of easy bruising who cannot show a single bruise at the time of the consultation, or one who agrees that there are fewer days with bruises than days without,11 is less likely to have a bleeding disorder. Thrombocytopenic purpura crop around the ankles, where venous pressure is highest, and are more likely to be perceived as ‘a rash’ than as bruises.
Drug history
A full list of all prescribed or over-the-counter medication (including herbal and other complementary medicines) taken by the individual should be compiled. Aspirin remains the most prevalent agent causing bleeding symptoms and abnormal platelet function test results: in addition to being prescribed widely for its antithrombotic effect, it is a component of many preparations on sale to the public. Some of these preparations have names that advertize the presence of aspirin (e.g. Aspro®) while others (e.g. Nurse Sykes’ Powders®) do not: a full list of such products is given in the British National Formulary.14 Other non-steroidal anti-inflammatory agents share the aspirin effect. Antibiotics, major tranquillizers and antidepressive agents may all be associated with bleeding via antiplatelet function effects. Platelet function testing should be performed first with the patient taking the drug, then 2 weeks after stopping, in order to demonstrate its effect.
Clinical examination
Musculoskeletal system
Joints should be examined for warmth, effusion, synovitis, reduced range of movement and misalignment. Muscle groups should be examined for wasting and contractures. These signs of cumulative damage due to hemarthrosis and intramuscular hematomas are characteristic of the hemophilias, but are also seen in rare disorders such as type 3 von Willebrand disease (vWD), deficiency or severe recessive disorders such as homozygous factor XIII, factor VII or factor X deficiency. Intramedullary hemorrhage of the long bones is a feature of afibrinogenemia and α2-antiplasmin deficiency, both very rare: it mimics lytic bone disease.15
On defining the pretest probability of a bleeding disorder
Using information from the history and examination the clinician can work out a broad pretest probability (e.g. low, moderate or high) that the patient has a clinical bleeding disorder. The accuracy and precision of the history and examination described above in defining this pretest probability have not been tested by methods that have provided such information in other contexts.13 Such studies are feasible and desirable in bleeding disorders, but even in their absence, a rational estimate of pretest probability is a crucial step towards interpreting the results of laboratory testing. Without it, tests of hemostasis can be frankly misleading.
Laboratory investigation of hemostasis
Tests of primary hemostasis
Screening tests
The platelet count
The validity of the platelet count accordingly depends on instrument standardization, calibration and quality control: details of these procedures can be found elsewhere.15 Because instruments count particles by size, blast cell fragments (in acute leukemia) or schistocytic red cells (in thrombotic thrombocytopenic purpura) may lead to overestimation, and large platelets (in immune thrombocytopenia or myelofibrosis) to underestimation, of the true platelet count.
Normal and abnormal platelet counts. The normal (‘Gaussian’) reference range for the concentration of platelets in venous blood (’the platelet count’) is 150–400 × 109/l. By definition, 5% of normal individuals have platelet counts outside this range. To regard and investigate asymptomatic individuals with isolated, stable, mild thrombocytopenia (100–150 × 109/l) as if they had a disease may be to confound ‘Gaussian’ and ‘diagnostic’ concepts of normality.1 However, evidence to justify abandoning this seemingly unproductive practice is lacking.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


