As with all proteins that have been strongly conserved throughout evolution, the tertiary structure of globins is constant; virtually all globins have seven or eight helical regions (depending on the chain) (see Fig. 11-2). Mutations that disrupt this tertiary structure invariably have pathological consequences. In addition, mutations that substitute a highly conserved amino acid or that replace one of the nonpolar residues, which form the hydrophobic shell that excludes water from the interior of the molecule, are likely to cause a hemoglobinopathy (see Fig. 11-2). Like all proteins, globin has sensitive areas, in which mutations cannot occur without affecting function, and insensitive areas, in which variations are more freely tolerated.
The Globin Genes
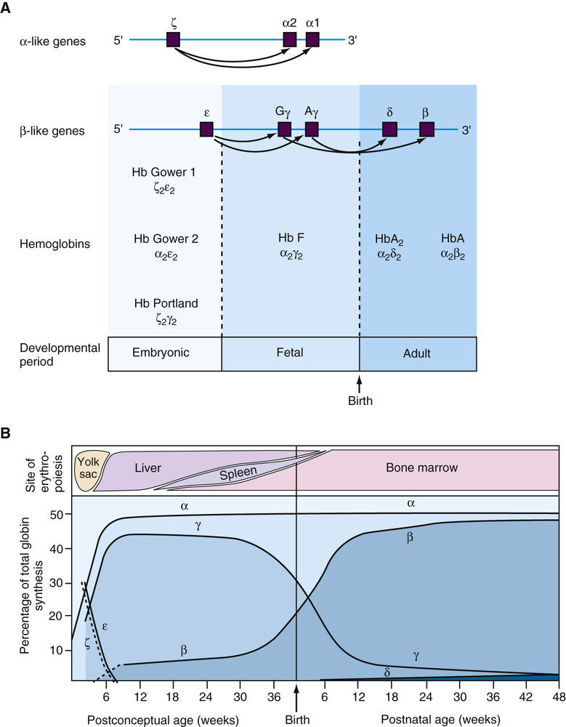
In addition to Hb A, with its α2β2 structure, there are five other normal human hemoglobins, each of which has a tetrameric structure like that of Hb A in consisting of two α or α-like chains and two non-α chains (Fig. 11-3A). The genes for the α and α-like chains are clustered in a tandem arrangement on chromosome 16. Note that there are two identical α-globin genes, designated α1 and α2, on each homologue. The β- and β-like globin genes, located on chromosome 11, are close family members that, as described in Chapter 3, undoubtedly arose from a common ancestral gene (see Fig. 11-3A). Illustrating this close evolutionary relationship, the β- and δ-globins differ in only 10 of their 146 amino acids.
Developmental Expression of Globin Genes and Globin Switching
The expression of the various globin genes changes during development, a process referred to as globin switching (see Fig. 11-3B). Note that the genes in the α- and β-globin clusters are arranged in the same transcriptional orientation and, remarkably, the genes in each cluster are situated in the same order in which they are expressed during development. The temporal switches of globin synthesis are accompanied by changes in the principal site of erythropoiesis (see Fig. 11-3B). Thus the three embryonic globins are made in the yolk sac from the third to eighth weeks of gestation, but at approximately the fifth week, hematopoiesis begins to move from the yolk sac to the fetal liver. Hb F (α2γ2), the predominant hemoglobin throughout fetal life, constitutes approximately 70% of total hemoglobin at birth. In adults, however, Hb F represents less than a few percent of the total hemoglobin, although this can vary from less than 1% to approximately 5% in different individuals.
β-chain synthesis becomes significant near the time of birth, and by 3 months of age, almost all hemoglobin is of the adult type, Hb A (α2β2) (see Fig. 11-3B). In diseases due to mutations that decrease the abundance of β-globin, such as β-thalassemia (see later section), strategies to increase the normally small amount of γ-globin (and therefore of Hb F (α2γ2)) produced in adults are proving to be successful in ameliorating the disorder (see Chapter 13).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


