Chapter 27 Heme Metabolism
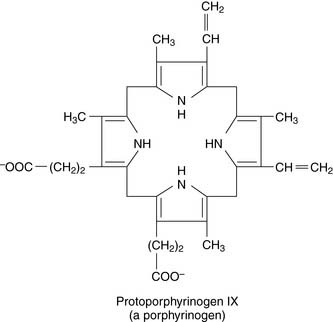
Porphyrinogens are prone to nonenzymatic oxidation to the corresponding porphyrins.
Bone marrow and liver are the most important sites of heme synthesis
The second most important site is the liver because of its high content of cytochrome P-450, which accounts for 65% of the total heme in the liver (see Chapter 30). P-450 enzymes are concerned with the inactivation of drugs and other foreign molecules, and the transcription of their genes is stimulated by many drugs. These enzymes have far shorter half-lives than does hemoglobin. Therefore hepatic heme synthesis is quite productive, accounting for approximately 15% of the total.
Heme is synthesized from succinyl-coenzyme A and glycine
Heme biosynthesis starts with the formation of Δ-aminolevulinate from succinyl-coenzyme A (succinyl-CoA) and glycine, catalyzed by the heme-containing, vitamin B6–dependent enzyme δ-aminolevulinate (ALA) synthase. This is the committed step of the pathway shown in Figure 27.1.
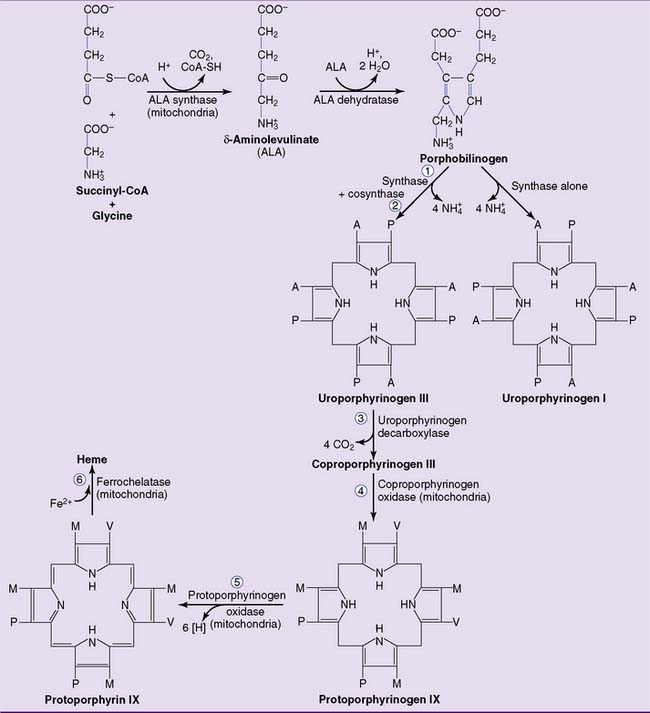
Figure 27.1 Pathway of heme biosynthesis. The numbered reactions refer to numbers listed in Table 27.1. A, Acetate (carboxymethyl) group; M, methyl group; P, propionate (carboxyethyl) group; V, vinyl group.
The second reaction, catalyzed by ALA dehydratase, forms the pyrrole ring of porphobilinogen. The third enzyme, uroporphyrinogen I synthase (also called porphobilinogen deaminase), links together four molecules of porphobilinogen. Left on its own, the synthase produces uroporphyrinogen I. In the cell, however, a second protein, uroporphyrinogen III cosynthase, channels the reaction into the formation of the isomer uroporphyrinogen III. All naturally occurring porphyrins, including heme, belong to the III series. Uroporphyrinogen III is processed to heme by the reactions shown in Figure 27.1.
Porphyrias are caused by deficiencies of heme-synthesizing enzymes
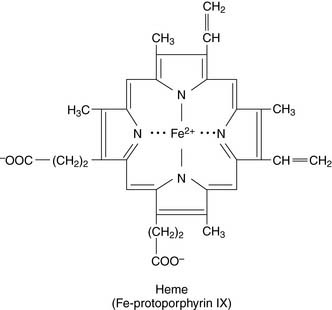
The porphyrias (Table 27.1) are caused by a partial deficiency of one of the heme-synthesizing enzymes other than ALA synthase. Any one of the numbered enzymes shown in Figure 27.1 can be affected. A complete deficiency would be fatal, and the offending mutations are generally expressed as autosomal dominant traits; affected heterozygotes have 50% of the normal enzyme activity. Clinically, we can distinguish between hepatic porphyrias and erythropoietic porphyrias.