FIGURE 16-1. Relationship between platelets and the clotting cascade in the generation of a stabilized fibrin clot. 5HT = serotonin; ADP = adenosine diphosphate; PF4 = platelet factor 4; TxA2 = thromboxane A2.
Once adhesion occurs, platelets change shape and activation occurs. Substances such as collagen, ADP, thrombin, and thromboxane A2 (TxA2) stimulate the change in platelet shape and cause platelets to release their contents including ADP, serotonin (5HT), platelet factor 3 (PF3), and platelet factor 4 (PF4).2 Following platelet adhesion and activation, platelet aggregation completes the formation of the hemostatic plug. This process is mediated by glycoprotein IIb/IIIa receptors on the platelet surface with fibrinogen acting as the primary binding ligand bridging between platelets. Platelets have numerous Gp IIb/IIIa binding sites, which are an attractive option for antiplatelet drug therapy.2,4
ADP and TxA2 recruit additional platelets, which aggregate to the platelets that are already bound to the subendothelial tissues. In addition to promoting aggregation, TxA2, 5HT, and other substances are potent vasoconstrictors that limit blood flow to the damaged site. When vascular damage is minimal, the vasoconstriction and platelet aggregation (formation of a platelet plug) may be sufficient to limit bleeding.
However, the platelet plug is not stable and can be dislodged. To form a more permanent hemostatic plug, the clotting system must be stimulated. By releasing PF3, platelets initiate the clotting cascade and concentrate activated clotting factors at the site of vascular (endothelial) injury.
Prostaglandins (PGs) play an important role in platelet function. Figure 16-2 displays a simplified version of the complex arachidonic acid pathways that occur in platelets and on the vascular endothelium. Thromboxane A2, a potent stimulator of platelet aggregation and vasoconstriction, is formed in platelets. In contrast, prostacyclin (PG2), produced by endothelial cells lining the vessel luminal surface, is a potent inhibitor of platelet aggregation and a potent vasodilator that limits excessive platelet aggregation.
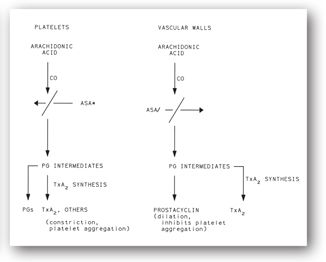
FIGURE 16-2. Formation of thromboxane A2 (TxA2), prostaglandins (PGs), and prostacyclin in platelets and vascular endothelial cells. CO = cyclooxygenase; ASA* = low-dose, irreversible, inactivation of platelet cyclooxygenase; ASA/ = high-dose inactivation of platelet cyclooxygenase.
Cyclooxygenase and PG2 are clinically important. An aspirin dose of 80 mg/day acetylates and irreversibly inhibits cyclooxygenase in the platelet. Platelets are rendered incapable of forming arachidonic acid and PGs. This effect of low-dose aspirin lasts for the lifespan of the exposed platelets (up to 12 days).
Vascular endothelial cells also contain cyclooxygenase, which converts arachidonic acid to PG2. Aspirin in high doses inhibits the production of PG2. However, because the vascular endothelium can regenerate PG2, aspirin’s effect is much shorter here than on platelets. Thus, aspirin’s effect at high doses may both inhibit platelet aggregation and block the aggregation inhibitor PG2. This phenomenon is the rationale for using low doses of aspirin 80–325 mg/day to help prevent myocardial infarction.
In summary, a complex interaction between the platelet and blood vessel wall maintains hemostasis. Once platelet adhesion occurs, the clotting cascade may become activated. After thrombin and fibrin are generated, the platelet plug becomes stabilized with insoluble fibrin at the site of vascular injury.
Coagulation Cascade
The ultimate goal of the coagulation cascade (Figure 16-3) is to generate fibrin from thrombin. Fibrin forms an insoluble mesh surrounding the platelet plug. Platelets concentrate activated clotting factors at the site of vascular injury.
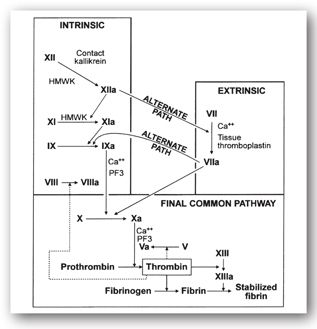
FIGURE 16-3. Coagulation cascade. Dotted lines indicate thrombin’s feedback action, which modifies factors V and VIII. HMWK = high molecular weight kininogen. (Reproduced, with permission, from reference 5.)
The nomenclature and half-lives for the coagulation proteins are shown in Table 16-1. The coagulation cascade is typically divided into the intrinsic, extrinsic, and common pathways: the intrinsic and extrinsic pathways provide different routes to generate factor X while the common pathway results in thrombin formation. Coagulation is initiated by vascular injury or damage that exposes blood to tissue factor (TF), which then binds to factor VII at the start of the extrinsic pathway. The binding of TF to factor VII activates the latter to VIIa. The complex formed by TF and factor VIIa can then activate factor X to Xa at the start of the common pathway. Alternatively, the TF-factor VIIa complex can first convert factor IX to factor IXa, with factor VIIIa as a cofactor, which is part of the intrinsic pathway. Factor IXa can then activate factor X into Xa; thus, both the intrinsic and extrinsic pathways activate factor X in the final common pathway. Factor Xa with factor Va as a cofactor activates prothrombin (factor II) into thrombin (factor IIa). In the clotting cascade, thrombin not only converts fibrinogen into fibrin, but it can also convert factor XIII to factor XIIIa, which stabilizes the fibrin clot.4 In addition to the direct effects and feedback mechanisms of thrombin shown in Figure 16-3, thrombin also stimulates platelet aggregation and activates the fibrinolytic system.
Additional factors within the pathway. Factors such as calcium and vitamin K play an intricate role within the various pathways in the coagulation cascade. Calcium is essential for the platelet surface binding of several factors within the pathway. Vitamin K facilitates the calcium binding function of factors II, VII, IX, and X via carboxylation. These processes are critical in activating proteins within the pathway.
Inhibition of Coagulation
Mechanisms that limit coagulation include the natural inhibitors such as antithrombin (AT) and the vitamin K dependent proteins C and S, tissue factor pathway inhibitor (TFPI), and the fibrinolytic system. Endothelial cells produce several substances that have antithrombotic effects which may also activate the fibrinolytic system.4 Platelet aggregation is prevented by substances such as PG2 and nitric oxide, both of which are generated by the vessel wall.1,4 Generation of plasminogen activators also can limit platelet aggregation.1 Several medications can also inhibit coagulation by acting on platelets (aspirin, clopidogrel, prasugrel) or one or more clotting factors (warfarin, low molecular weight heparins [LMWHs], unfractionated heparin [UFH], dabigatran, rivaroxaban, apixaban, and direct thrombin inhibitors [DTIs]).
High concentrations of thrombin, in conjunction with thrombomodulin, activate protein C, which can then inactivate cofactors Va and VIIIa; thus, there is a negative feedback mechanism that will block further thrombin generation and subsequent steps in the coagulation cascade.8 Protein S serves as a cofactor for protein C. Antithrombin inactivates thrombin as well as factors IX, X, and XI, and this process can be hastened by heparin. Heparin and AT combine one-to-one, and the complex neutralizes the activated clotting factors and inhibits the coagulation cascade. Deficiencies in these natural inhibitors can result in increased generation of thrombin, which can lead to recurrent thromboembolic events often starting at a young age. Tissue factor pathway inhibitor impedes the binding of TF to factor VII, essentially inhibiting the extrinsic pathway (Figure 16-3). Unfractionated heparin and LMWHs can release TFPI from lipoproteins.4 The complex mechanisms that limit thrombus formation are shown in Figure 16-4.
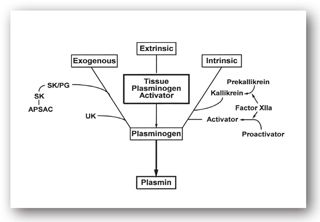
FIGURE 16-4. Exogenous, extrinsic, and intrinsic pathways for activation of plasminogen. APSAC = anistreplase; SK = streptokinase; SK/PG = streptokinase-plasminogen complex; UK = urokinase. (Reproduced, with permission, from reference 7.)
Clot Degradation
Fibrinolysis is the mechanism by which formed thrombi are lysed to prevent excessive clot formation and vascular occlusion. As discussed previously, fibrin is formed in the final common pathway of the clotting cascade. Tissue plasminogen activator (tPA) and urokinase plasminogen activator (uPA) activate plasminogen, which generates plasmin. Plasmin is the enzyme that eventually breaks down fibrin into fibrin degradation products (FDPs). Medications can either activate (e.g., streptokinase, alteplase, urokinase, reteplase, and tenecteplase) or inhibit (tranexamic acid, aminocaproic acid, and aprotinin) fibrinolysis.
TESTS TO EVALUATE HEMOSTASIS
For the purpose of discussion, bleeding and clotting disorders are organized by tests that assess
- Platelets
- Coagulation
- Clot degradation
- Hypercoagulable states
Tests to assess platelets include platelet count, volume (mean platelet volume [MPV]), function (e.g., bleeding time [BT] and platelet aggregation), and others. Thrombin time (TT), reptilase time, prothrombin time (PT)/international normalized ratio (INR), activated partial thromboplastin time (aPTT), activated clotting time (ACT), fibrinogen assay, and others are laboratory tests that assess coagulation. Clot degradation is assessed with tests for FDPs and D-dimer. Fibrinolysis also is monitored with the euglobulin lysis test.
A hypercoagulable state workup may include activated protein C (APC) resistance and the factor V Leiden mutation, anticardiolipin antibody, antiphospholipid antibody, antiplasmin, AT, C-reactive protein, heparin neutralization, homocystine, lipoprotein, plasminogen, plasminogen activator inhibitor 1 (PAI1), platelet hyperaggregation, proteins C and S, prothrombin G20210A mutation, reptilase time, and TT. These tests are often performed in panels since the presence of more than one predisposition to thrombosis further increases the risk for thrombosis.8
In addition, general hematological values such as hemoglobin (Hgb), hematocrit (Hct), red blood cell (RBC) count, and white blood cell (WBC) count, as well as urinalysis and stool guaiac tests may be important to obtain when evaluating blood and coagulation disorders; some of these tests are further discussed in Chapter 15: Hematology: Red and White Blood Cell Tests. Table 16-2 is a summary of common tests used to evaluate bleeding disorders and monitor anticoagulant therapy.
aPTT= activated partial thromboplastin time; BT= bleeding time; ITP= idiopathic thrombocytopenic purpura; PT/INR= prothrombin time/international normalized ratio; TTP= thrombotic thrombocytopenic purpura; vWF= von Willebrand factor; WNL= within normal limits.
aItalic type indicates most useful diagnostic or therapeutic tests.
bSignificant thrombocytopenia may occur as a heparin side effect in 1% to 5% of patients.
Platelet Tests
Platelet Count
Normal range: 150,000–450,000/µL
The only test to determine the number or concentration of platelets in a blood sample is the platelet count, through either manual (rarely done) or automated methods. Interferences with platelet counts include RBC fragments, platelet clumping, and platelet satellitism (platelet adherence to WBCs). Automated platelet counts are performed on anticoagulated whole blood. Most instrumentation that performs hematological profiles provides platelet counts. Platelets and RBCs are passed through an aperture generating an electric pulse with a magnitude related to the size of the cell/particle. The pulses are counted, and the platelets are separated from the RBCs by size providing the platelet count and MPV as well as the RBC count and mean corpuscular volume (MCV).
Thrombocythemia. An abnormal platelet count can have many causes. Thrombocythemia, also known as thrombocytosis or elevated platelet count, may be caused by several factors including
- Stress
- Infection
- Splenectomy
- Trauma
- Asphyxiation
- Rheumatoid arthritis
- Iron-deficiency anemia
- Posthemorrhagic anemia
- Cirrhosis
- Chronic pancreatitis
- Tuberculosis
- Occult malignancy
- Recovery from bone marrow suppression
Values of 500,000–800,000/µL are not uncommon. Thrombocythemia may be seen with any of the chronic myeloproliferative neoplasms, essential thrombocythemia, polycythemia vera, chronic myelogenous leukemia, or idiopathic myelofibrosis. Clinical consequences of thrombocythemia include thrombosis, hemorrhage, and microcirculatory disturbances. Thrombotic events may be either arterial or venous and include cerebrovascular accidents, myocardial infarction, deep venous thrombosis, pulmonary embolism, and intra-abdominal (portal and hepatic) vein thrombosis. Hemorrhagic complications usually involve the skin and/or mucous membranes, which include ecchymosis, epistaxis, and menorrhagia. Microcirculatory disturbances, such as headache, paresthesias, and erythromelalgia, may be due to microthrombi, which results in occlusion and ischemia. Additionally, patients with thrombocythemia may have abnormalities in platelet function studies, which can manifest as bleeding problems.
Thrombocytopenia. Mucosal and/or cutaneous bleeding is the most common clinical consequence of thrombocytopenia; however, patients with only modest decreases in platelet counts may be asymptomatic. (See Minicase 1.) When the platelet count falls below 20,000/µL, the patient is at risk of spontaneous bleeding. Therefore, platelet transfusions are often initiated. Bleeding may occur at higher platelet counts (e.g., 50,000/µL) if trauma occurs. The most common cause of death in a patient with severe thrombocytopenia is central nervous system (CNS) bleeding such as intracranial hemorrhage.
Bleeding Disorders
HELEN M., A 56-YEAR-OLD FEMALE, was hospitalized with a longstanding history of cirrhosis. Physical examination revealed a cachectic-appearing female with distended abdomen, petechial hemorrhages, and asterixis. Helen M. was taking lisinopril, metoprolol, pravastatin, ranitidine, allopurinol, and MVI. She denied taking over-the-counter medications.
The following laboratory parameters were determined for Helen M.:
| LABORATORY STUDY | NORMAL RESULTS | PATIENT’S TEST RESULTS |
| PT | 10–13 sec | 14.8 sec |
| INR | Varies around 0.9–1.1 | 1.49 |
| aPTT | 21–45 sec | 54 sec |
| CBC | ||
| Hgb | 12.3–15.3 g/dL | 11 g/dL |
| Hct | 42% to 52% | 33% |
| Platelet count | 140,000–440,000/µL | 87,000/µL |
| MPV | 7–11 fL | 14 fL |
| AST | 8–42 International Units/L | 85 International Units/L |
| ALT | 3–30 International Units/L | 25 International Units/L |
| Albumin | 3.5–5 g/dL | 1.4 g/dL |
Question: What specific test(s) were performed to assess the pertinent patient findings from the history and physical examination? How might these tests relate to normal hemostasis?
Discussion: Preliminary screening was done using the PT/INR and aPTT; they were used as a preliminary screening for Helen M. Her elevations above baseline are consistent with coagulopathies common in patients with liver dysfunction. This increase occurs because of a diminished ability of the liver to manufacture clotting factors.
Platelet count determination and general hematologic values (Hgb and Hct) are consistent with physical examination findings (petechial hemorrhages), revealing Helen M. to be moderately thrombocytopenic and mildly anemic. As expected, the lowered platelet count and higher platelet volume demonstrate the inverse relationship that typically exists when thrombocytopenia occurs. The lowered platelet count could be due to splenic sequestration of platelets due to splenomegaly in association with liver cirrhosis. However, the physical examination did not describe splenomegaly, and the MPV would not be elevated in this situation. Liver function and albumin tests suggest that cirrhosis is the etiology of Helen M.’s petechial hemorrhages. There could be a consumptive component to the coagulopathy as well, which would explain the moderate thrombocytopenia with an increased MPV due to platelet destruction. Table 16-2 summarizes common tests to evaluate bleeding disorders and monitor anticoagulant therapy.
With Helen M., history is important. Both allopurinol and ranitidine have been associated with thrombocytopenia, which could also account for petechial hemorrhages. Helen M. denies using aspirin or NSAIDs, but many patients unknowingly ingest over-the-counter products, such as analgesics and cold preparations. Aspirin and NSAIDs interfere with platelet aggregation and can prolong BT.
Numerous drugs have been associated with thrombocytopenia (Table 16-3). However, heparin and antineoplastics are the most common ones implicated. Thrombocytopenia is also common with radiation therapy. Many drugs associated with thrombocytopenia alter platelet antigens resulting in the formation of antibodies to platelets (e.g., heparin, penicillin, and gold). Several diseases, such as thrombotic thrombocytopenic purpura (TTP), idiopathic thrombocytopenic purpura (ITP), disseminated intravascular coagulation (DIC), and hemolytic-uremic syndrome, result in rapid destruction of platelets. Other causes of thrombocytopenia include viral infections; pernicious, aplastic, and folate/B12-deficiency anemias; complications of pregnancy; massive blood transfusions; exposure to dichlorodiphenyltrichloroethane (DDT); and human immunodeficiency virus (HIV) infections.
aSome drugs listed cause an idiosyncratic, drug-induced thrombocytopenia while others induce formation of antiplatelet antibodies (gold, penicillins, and heparin). Exceptions include amrinone, antineoplastics, and possibly amphotericin B, which cause a direct toxic effect that may be dose related.
Heparin-induced thrombocytopenia (HIT) is an antibody-mediated adverse reaction to heparin, occurring in 0.2% to 5% of all heparin-treated adults, which may cause venous and arterial thrombosis.9 Specifically, this is due to the development of IgG antibodies that bind to the heparin PF4 complex. Patients receiving UFH are generally at a higher risk of developing HIT than patients receiving LMWH. Low molecular weight heparin does not bind as well as UFH to PF4, which is thought to be due to the smaller size of LMWH compared to UFH. Therefore, the heparin-PF4 complex is less likely to form with LMWH, and there are less IgG antibodies generated. The frequency or risk of HIT is influenced by certain factors such as heparin preparation, route, dose, and duration of heparin therapy, patient population, gender, and previous history of heparin exposure.10,11 The animal source of heparin may also play a role in determining who develops HIT; bovine UFH seems to carry a higher risk compared to porcine UFH.11
The 4Ts score is a clinical prediction tool to help physicians determine the probability of their patient having HIT. This tool looks at thrombocytopenia, timing of platelet count fall or thrombosis, thrombosis or other clinical sequelae, and other causes for thrombocytopenia; a score of 0–2 is assigned for each of the four parts based on specific patient characteristics to determine the probability of HIT occurring in that particular patient.10 Heparin-induced thrombocytopenia is manifested both by clinical and serological features, and diagnosis of HIT is usually made when antibody formation is detected by an in vitro assay plus one or more of the following: unexplained decrease in platelet count (usually ≥30%, even if the nadir remains above 150 × 109/L), venous or arterial thrombosis, limb gangrene, necrotizing skin lesions at the heparin injection site, or acute anaphylactoid reactions occurring after intravenous (IV) heparin bolus administration.10
There are two types of tests to help diagnose HIT: the enzyme-linked immunosorbent assay (ELISA), which identifies anti-PF4/heparin antibodies and functional assays, such as the C-serotonin release assay (SRA) or the heparin-induced platelet activation (HIPA) assay, both of which detect antibodies that induce heparin-dependent platelet activation.9-11 The ELISA test has high sensitivity and wide availability, with a relatively rapid turnaround time compared to the functional assays, which makes it a good screening test. However, the ELISA test has limited specificity so there may be false-positive results, especially in patients with antiphospholipid syndrome or systemic lupus erythematosus.9,11 In contrast, the functional assays have high specificity, which are useful for confirming a positive ELISA test but are technically difficult and require the use of radioactivity and/or donor platelets.9,11
The typical onset for HIT is 5–10 days following the start of heparin; however, onsets occurring either earlier or later than this have been reported. Rapid-onset HIT occurs when platelet counts fall within 24 hours of heparin initiation, which is typically due to repeated heparin exposure within the past 100 days, and thus patients still have circulating HIT antibodies. Delayed-onset HIT, where thrombocytopenia occurs several days after discontinuation of heparin, has also been reported and is associated with DIC.9,12-17 Platelet counts should be checked in patients receiving UFH or LMWH if the clinician deems the risk of HIT >1%; in these cases, recommendations are for platelet counts to be done every 2–3 days from days 4–14, or when heparin is stopped.10 If the risk of HIT is <1%, then platelet monitoring is not recommended.10 For patients who received heparin within the past 100 days, platelets should be checked at baseline and then within 24 hours of starting heparin.10 Table 16-4 outlines the patient characteristics associated with the risk of developing HIT.
HIT = heparin-induced thrombocytopenia; LMWH = low molecular weight heparin; UFH = unfractionated heparin.
If HIT is suspected and/or confirmed, UFH and/or LMWH should be discontinued. Direct thrombin inhibitors, such as argatroban, lepirudin, or bivalirudin, can be used instead of UFH or LMWH and are FDA-approved for use in HIT. Fondaparinux is a synthetic, indirect inhibitor of factor Xa. Although it is not FDA-approved for use in patients with HIT, there have been reports of successfully using fondaparinux as an alternative anticoagulant in the HIT population; however, there are also reports of fondaparinux-associated HIT or complications from using fondaparinux in patients with HIT.18-30,31-37 The 9th edition of the American College of Chest Physicians Evidence-Based Clinical Practice Guidelines recommends the use of fondaparinux in HIT as a second-line agent to other DTIs in hospitalized patients or as a first-line parenteral agent in patients who develop an acute thrombosis unrelated to HIT as a bridge until warfarin therapy can be used.10 In patients who require warfarin for longer term anticoagulation, it is recommended to wait to start warfarin until platelets have recovered to at least 150 × 109/L and to start at low doses.10
Mean Platelet Volume
Normal range: 7–11 fL (varies with laboratory)
Mean platelet volume (MPV), the relationship between platelet size and count, is most likely to be used by clinicians in assessing disturbances of platelet production. Mean platelet volume is useful in distinguishing between hypoproductive and hyperdestructive causes of thrombocytopenia (Figure 16-5). Despite the widespread availability of this platelet index, many clinicians do not use it in clinical decision-making. In the past, this disuse was attributed to difficulties with the laboratory measurement of indices.
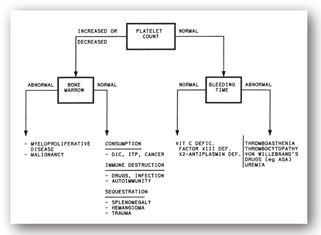
FIGURE 16-5. Assessment of abnormalities of homeostasis based on platelet count, bone marrow exam, and BT.
Many laboratories routinely report the MPV as part of the complete blood count (CBC), especially if a differential is requested. In general, lower platelet counts are common with higher platelet volumes, as an inverse relationship exists between the platelet count and the MPV. This inverse relationship correlates with platelet production within the bone marrow. Although MPV is most valuable in distinguishing hypoproductive from hyperdestructive causes of thrombocytopenia, a definitive diagnosis cannot be made based on MPV alone. In thrombocytopenia, an elevated MPV suggests no problem with platelet production, when in fact, production is reflexively increased. Conversely, a normal or low MPV suggests impaired thrombopoiesis. Determination of MPV requires a blood collection tube containing an anticoagulant. Usually, such tubes contain the anticoagulant ethylenediamine tetraacetic acid (EDTA), which causes an inflation of the MPV.38
Currently, MPV is not widely used but may evolve into a valuable screening test for the disorders listed in Table 16-5. Interesting data relating MPV to these disorders continues to surface. For example, the MPV is often elevated at the time of myocardial infarction, although it is not specific enough to be of diagnostic value. Studies have suggested though that a high MPV at 6 months postinfarction may be a predictor of reinfarction. Mean platelet volume is altered in the presence of numerous other medical conditions (Table 16-5).39,40 For example, a fall in MPV is common in patients with enlarged spleens (hypersplenism) due to preferential sequestering of larger platelets within the spleen. An increase in MPV is seen during the third trimester of pregnancy in preeclamptic patients, where an increase in platelet size results from increased platelet consumption.41 The MPV is also elevated in hyperthyroid patients but declines to normal as they become euthyroid. In contrast, hypothyroid patients often have a high platelet count and a low MPV.42,43
aReference ranges for MPV vary among laboratories and are inversely proportional to the platelet count.
The inverse relationship of a high MPV and a low platelet count is demonstrated in other conditions including respiratory disease, renal failure, and sepsis.44-46 Unlike most other conditions that demonstrate the inverse relationship of MPV and platelet count, both are low in HIV infection. These decreases suggest an impairment of synthesis and maturation of megakaryocytes as well as enhanced platelet destruction in the bloodstream.47 Administration of erythropoietin stimulates megakaryocyte cell line production that leads to an increase in MPV. Thrombopoietin probably causes the same effect.48-51 The role of thrombopoietin has lead to advances in the treatment of thrombocytopenia caused by deficient production of platelets (e.g., in patients undergoing bone marrow transplantation or cancer chemotherapy).51
Abnormalities of platelet function may be either inherited or acquired. Bleeding as a result of an inherited versus acquired abnormality may be difficult to prove. Common bleeding sites in patients with inherited disorders of platelet function include ecchymosis of the skin, epistaxis, gingival bleeding, and menorrhagia. Less common bleeding disorders are gastrointestinal hemorrhage and hematuria. Hematomas and hemarthroses (the predominant sites of bleeding in patients with inherited familial clotting disorders) are rare, except after trauma.52
Although the sites of bleeding are predictable, the severity is not predictable in patients with inherited disorders of platelet function.52 Unfortunately, the risk of bleeding and bleeding patterns in patients with acquired platelet dysfunction are less predictable and more difficult to distinguish. Because both inherited and acquired etiologies increase the risk of bleeding, patients overtly bleeding without a clear cause or without an invasive procedure should be suspect for one of these platelet function disorders.52
Bleeding time (normal range: 2–9 minutes). The bleeding time (BT) is a measure of platelet function, but this test is neither specific, nor sensitive; thus, it does not help differentiate among the types of problems seen in disorders of primary hemostasis, such as von Willebrand disease and platelet function defects. This would account for its declining use and elimination by some institutional clinical laboratories. Additionally, the test is invasive and must be performed by a trained healthcare worker. To perform the test, small cuts are made on the forearm of the patient, and the time it takes to stop bleeding is measured.1 Several factors can prolong the BT including thrombocytopenia, certain medications, and conditions such as uremia and macroglobulinemia. Most acquired disorders affecting BT are related to medications that decrease platelet numbers or reduce platelet function. These include aspirin, thienopyridines (ticlopidine, clopidogrel, prasugrel), GPIIb/IIIa inhibitors (abciximab, eptifibatide, tirofiban), and phosphodiesterase inhibitors (dipyridamole).
Aspirin irreversibly acetylates cyclooxygenase (Figure 16-2). Platelets cannot synthesize new cyclooxygenase, and the effect of aspirin persists for the lifespan of the exposed platelets. The prolongation of BT caused by aspirin is somewhat shorter than the platelet lifespan (8–12 days), because platelets formed after aspirin exposure are not affected. Heparin and warfarin may also prolong BT but not by affecting platelet aggregation. Other drugs that may prolong BT are listed in Table 16-6. While nonacetylated salicylates do not impair platelet aggregation or affect BT, all other nonsteroidal anti-inflammatory drugs (NSAIDs) reversibly inhibit platelet cyclooxygenase and inhibit aggregation as long as the drug remains in the plasma. Therefore, the clinician can estimate the time it takes for BT to normalize after withdrawing these drugs by knowing their respective half-lives (i.e., 100% elimination of NSAIDs from the blood stream occurs after five half-lives have elapsed). Although BT is influenced by some drugs, it is not used to monitor drug therapy. The increase in BT caused by aspirin may have beneficial effects in the treatment and prevention of cardiovascular disease. The proven value of aspirin for acute treatment of myocardial infarction and secondary prevention of cardiovascular disease is well established; however, the use of aspirin for primary prevention of cardiovascular disease remains an individual clinical judgment.
Source: Adapted from reference 53.
Platelet aggregation. With the many drawbacks of the BT, there was a need for a test that could aid in the diagnosis of defects in platelet function. This is especially true when considering the interpatient variability seen when taking clopidogrel. Adverse events may occur if patients do not respond to this medication. The ability of platelets to aggregate is most commonly measured by preparing a specimen of platelet-rich plasma and warming it to 98.6°F (37°C) with constant stirring. This test is performed with an aggregometer that measures light transmission through a sample of platelets in suspension. After a baseline reading is obtained, a platelet-aggregating agonist (e.g., epinephrine, collagen, ADP, or arachidonic acid) is added. As platelets aggregate, more light passes through the sample. The change in optical density can be measured photometrically and recorded as an aggregation curve, which is then printed on a plotter. Although this light transmittance aggregometry (LTA) is the gold standard in platelet function analysis, it is expensive and requires specially trained personnel to run the test.
Interpretation of platelet aggregation tests involves a comparison of the patient’s curves with the corresponding curves of a normal control. To eliminate the optical problems of turbidity with lipemic plasma, the patient and the normal control should be fasting. Patients should not take medications that affect platelet aggregation (e.g., aspirin, NSAIDs, clopidogrel, prasugrel) for approximately 7–14 days prior to the test because they may interfere with test results.
Novel point-of-care technologies, including the Platelet Function Analyzer (PFA-100, DadeBehring) and the Rapid Platelet Function Assay (RPFA) (Verify Now™ Accumetrics, Inc.), allow for rapid and meaningful evaluation of platelet function, although major differences between different devices do exist. The PFA-100 is not as sensitive to clopidogrel while the RPFA is sensitive to the effects of GPIIB/IIIa antagonists.54
Other Platelet Tests
The measurement of platelet-specific substances, such as PF4 and beta-thromboglobulin, can now be performed by radioimmunoassay or enzyme immunoassay. High concentrations of these substances may be observed with CAD, acute myocardial infarction (AMI), and thrombosis, where platelet lifespan is reduced. Since numerous drugs can potentially cause thrombocytopenia, detection of antibodies directed by specific drugs against platelets may help to determine the culprit. Platelet survival can be measured by injecting radioisotopes that label the platelets. Serial samples can then determine platelet survival, which is normally 8–12 days.
Coagulation Tests
Coagulation tests are useful in the identification of deficiencies of coagulation factors responsible for bleeding as well as thrombotic disorders. The most commonly performed tests, including the PT, INR, aPTT, and ACT, also are used to monitor anticoagulant therapy. Numerous, high-precision automated laboratory methods are available to perform these tests. However, an overall lack of standardization across coagulation testing can lead to considerable variation in test results and their interpretation. Normal and therapeutic ranges established for one test method are not necessarily interchangeable with other methods, especially when differences in endpoint detection or reagents exist. Therefore, it is important to interpret test results based on the specific performance characteristics of the method used to analyze samples.
Careful attention to blood collection technique and sample processing—as well as laboratory quality control—is critical for reliable coagulation test results. Blood is collected in syringes or vacuum tubes that contain heparin, EDTA, or sodium citrate. Because heparin and EDTA interfere with several clotting factors, only sodium citrate is used for coagulation and platelet tests. Errors in coagulation can be large unless quality assurance is strict with specimen collection, reagents, controls, and equipment. Factors that promote clotting and interfere with coagulation studies include
- Tissue trauma (searching for a vein)
- Prolonged use of tourniquet
- Small-bore needles
- Vacuum tubes
- Heparin contamination from indwelling catheters
- Slow blood filling into collection tube
Coagulation studies may be used to assess certain bleeding disorders such as hemophilia A (factor VIII deficiency) or hemophilia B (factor IX deficiency). These deficiencies, which are inherited sex-linked recessive traits, primarily affect males and cause over 90% of hemophilia cases. Other bleeding disorders include von Willebrand disease, the most common hereditary bleeding disorder, and deficiencies in fibrinogen or factors II, V, VII, X, XI, XIII, and/or a combination of these factors.
Patients with thrombotic disorders may have their hypercoagulability evaluated with specific assays for
- Antithrombin
- Protein C
- Protein S
- Prothrombin G20210A mutation (factor V Leiden)
- APC resistance mutation
Normal reference ranges for AT and proteins C and S tests are often reported as a percent of normal activity, with 100% being the mean normal value. For AT, the normal activity level is 80% to 130%; for both proteins C and S, normal activity levels are 70% to 140%. As alluded to previously, deficiencies can result in frequent, recurrent thromboembolic events in patients with these disorders. Because these deficiencies are rare, their respective assays are not discussed here in detail. Acquired, transient deficiencies of any of these inhibitors may be observed during thrombotic states. Therefore, these parameters should not be assessed during the acute phase of thrombosis or while the patient is currently on anticoagulant therapy since a false-positive result may occur. It is recommended to test for AT, protein C, protein S, and APC resistance after the thrombosis has been resolved when the patient is off of heparin or warfarin for approximately 2 weeks; the test for prothrombin G20210A mutation is not affected by current anticoagulant therapy.55
Activated protein C resistance, due to the factor V Leiden mutation, is the most prevalent hereditary predisposition to venous thrombosis. It is present in 5% of the general Caucasian population and is less common or rare in other ethnic groups.55-58 It accounts for 20% of unselected patients with a first deep vein thrombosis and 50% of familial cases of thrombosis.59 Patients with the heterozygous form of factor V Leiden mutation are at four- to sevenfold higher risk of developing venous thromboembolism (VTE), while those with the homozygous form can be as high as 80-fold higher risk.56 APC usually prolongs the aPTT more than twofold in controls (normal persons) and less than twofold in affected individuals. Presence of lupus anticoagulants, lepirudin, argatroban and bivalirudin may cause inaccurate results in the commonly used aPTT clotting-time based assay but do not affect DNA-based tests.60,61
Prothrombin G20210A mutation is the second most common hereditary predisposition to venous thrombosis. DNA-based methods, such as the polymerase chain reaction (PCR)-based assay, are used to determine the presence or absence of a specific mutation at nucleoside position 20210 in the prothrombin gene. A normal test would show absence of the G20210A mutation. The test identifies individuals who have the G20210A mutation and reveals whether the affected individual is heterozygous or homozygous for the mutation; patients with the heterozygous form are at a two- to fourfold higher risk of venous thrombosis, while those with the homozygous form are at even higher risk.56
Prothrombin Time/International Normalized Ratio
Normal range for PT: 10–13 sec but varies based on reagent-instrument combinations; therapeutic range for INR depends on indication for anticoagulation; most indications: 2.0–3.0
The prothrombin time (PT), also called ProTime, test is used to assess the integrity of the extrinsic and common pathways (factors II, V, VII, X). The PT, based on Quick’s method first described in 1935, is determined by adding calcium and an activator (thromboplastin) containing both the TF and the phospholipid necessary to promote the activation of factor X by factor VIIa in the patient’s plasma.62 The time it takes for clot formation to occur after the addition of the thromboplastin and calcium is the PT. Deficiencies or inhibitors of extrinsic and/or common pathway clotting factors would result in a prolonged PT; however, it should be noted that the PT is more sensitive to deficiencies in the extrinsic pathway (factor VII) compared to the common pathway (factors V, X, II, and fibrinogen).63
Assay performance characteristics, standardization, and reporting. The PT is dependent on the thromboplastin source and test method used to detect clotting. Thromboplastin reagents are derived from animal or human sources and include recombinant products. Factor sensitivity is highly dependent on the source of the thromboplastin, and can exhibit variability between different lots of the same reagent. Some thromboplastin reagents are less sensitive to changes in factor activity. This means that it takes a more significant decrease in factor activity to produce a prolongation of the PT. Differences in reagent sensitivity, combined with the influence of endpoint detection, affect clotting time results both in the normal and therapeutic ranges. Large differences in factor sensitivity between comparative methods can result in conflicting interpretation of results, both in the assessment of factor deficiencies and adequacy of anticoagulation therapy.
Since PT results can vary widely depending on the thromboplastin source, a standardized reporting method has been used, which is known as the international normalized ratio (INR). The INR is calculated according to the following equation:
INR = (patient PT/mean normal PT)ISI
The international sensitivity index (ISI) expresses the sensitivity of the thromboplastin reagent compared to the World Health Organization (WHO) reference standard. The more sensitive or responsive the reagent, the lower the ISI; reagents with ISI values of <1.7 are recommended for use when monitoring patients on oral anticoagulant therapy.62 Theoretically, an INR result from one laboratory should be comparable to an INR result from a different laboratory, even though the PTs may be different. The citrate concentration also may affect the ISI determination of certain reagents, with higher citrate concentrations leading to higher INR results; using blood samples anticoagulated with 3.2% citrate, instead of higher concentrations, can help mitigate this problem.
There are other factors that may influence the PT. If heparin-sensitive thromboplastin reagents are used, falsely elevated PT/INR values may result. These inaccurate values might suggest sufficient anticoagulation with oral anticoagulation therapy and result in the premature discontinuation of heparin.
Although the INR system has greatly improved the standardization of the PT, one can still expect differences in INRs reported with two different methods, particularly in the upper therapeutic and supratherapeutic ranges. The greater the differences in the ISI values for two comparative methods, the more likely differences will be noted in the INR. Laboratories and anticoagulation clinics should review the performance characteristics of the PT method used to evaluate their specific patient populations and report changes in methods to healthcare professionals, particularly those monitoring anticoagulant therapy.
Heparin may also prolong PT since it affects factor II in the common pathway; the addition of a heparin neutralizing agent to the blood sample can blunt this effect at heparin concentrations up to 2 units/mL.63 However, at higher concentrations of heparin, whether due to higher doses of heparin or sample collection issues, the neutralizing agent may not be enough and the PT may be prolonged. These “crossover” effects may have to be considered when oral and parenteral anticoagulants are given concomitantly for several days to avoid premature discontinuation of the parenteral agent.
Monitoring warfarin therapy. Both the PT and INR may be reported when monitoring warfarin therapy, although clinically, only the INR is used to adjust therapy. Warfarin exerts its anticoagulant effects by interfering with the synthesis of vitamin K-dependent clotting factors (II, VII, IX, and X) and the natural anticoagulant proteins C, S, and Z. Specifically, warfarin inhibits vitamin K-reductase and vitamin K epoxide-reductase (VKOR), which blocks the activation of vitamin K to its reduced form. Reduced vitamin K is needed for the carboxylation of clotting precursors of factors II, VII, IX, and X. Noncarboxylated clotting factor precursors are nonfunctional, and thus an anticoagulated state is achieved.62 Warfarin is manufactured as a racemic mixture of (S)- and (R)-enantiomers; the S-enantiomer is more potent than the R-enantiomer at inhibiting VKOR, which is why the S-enantiomer is responsible for the majority of the anticoagulant effects of warfarin. The S-enantiomer is metabolized largely by CYP2C9 while the R-enantiomer is metabolized mostly by CYP1A2, and CYP3A4; other CYP enzymes are also involved in the metabolism of warfarin although to a lesser extent.
Current Antithrombotic Therapy and Prevention of Thrombosis: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines recommend an INR of 2.0–3.0 for most indications; exceptions include, but are not limited to, patients with mechanical prosthetic heart valves in the mitral position and patients with recurrent thromboembolic events with a therapeutic INR, where an INR of 2.5–3.5 is recommended.64-68 Results below the therapeutic range indicate that the patient is at increased risk for clotting, and warfarin doses may need to be increased. Results above the therapeutic range indicate the patient is at risk for bleeding and warfarin doses may need to be decreased. Numerous drugs, disease states, and other factors prolong the INR in patients receiving warfarin by various mechanisms of action (Table 16-7).
Pharmacogenomics and oral anticoagulant therapy. Genetic variability in the genes coding for CYP2C9, VKORC1 (vitamin K epoxide reductase complex subunit 1), and CYP4F2 can influence warfarin dosing by altering its pharmacokinetics and/or pharmacodynamics. CYP2C9 and VKORC1 have a larger influence compared to CYP4F2. Specifically, patients with CYP2C9*2 and CYP2C9*3 variations have a reduced clearance of the (S)-warfarin enantiomer, which results in lower maintenance dose requirements of warfarin, increased risk of bleeding, and a possible longer time to achieve a stable dosing regimen.71 Two main haplotypes of VKORC1, low-dose haplotype group A, seen predominantly in Asian patients, and high-dose haplotype group B, seen predominantly in African-American patients, contribute to the interindividual variability of warfarin dosing. The specific single nucleotide polymorphisms (SNPs) involved are the -1639G>A and 1173C>T.
Patients with the AA genotype (predominately Asians) require lower doses of warfarin compared to Caucasians, while patients with the GG genotype (predominantly African-Americans) require higher doses compared to Caucasians.71 The CYP4F2 enzyme normally plays a role in the conversion of vitamin K to vitamin KH2, which is needed to carboxylate the clotting factor precursors; patients with a polymorphism in the 433Met allele of the CYP4F2 gene will have greater vitamin K availability leading to higher warfarin dose requirements.71 Unlike CYP2C9 polymorphisms, neither VKORC1 nor CYP4F2 have been associated with increased bleeding risks or prolonged time to achieving a stable dose of warfarin. The Hispanic population is underrepresented in warfarin pharmacogenomic studies, so until further data becomes available, Hispanics and non-Hispanic Caucasians are likely considered similar in terms of pharmacogenetic outcomes related to warfarin.71
Four manufacturers are currently marketing their warfarin pharmacogenomics testing devices; each one tests for the CYP2C9*2 and CYP2C9*3, as well as either the VKORC1-1639G/A or the VKORC1 1173C/T SNPs.71 The product labeling for Coumadin® (warfarin) has been updated to include information about the potential impact of pharmacogenomics on the dosing of this medication as well as a pharmacogenetics dosing table, which may help clinicians select an initial dose of warfarin if genetic information is known, specifically in regard to CYP2C9 and VKORC1.72 For clinicians who do utilize genetic testing for their patients on warfarin, they can use either the dosing table provided in the package insert or a dosing algorithm to estimate a starting dose for their patient. Algorithms take into account not only the results of genetic testing, but also other factors such as age, body size, smoking status, use of other medications like amiodarone, other disease states, and vitamin K intake. Subsequent dosing changes should be made based on results of the INR test. There are several barriers to widespread adoption of pharmacogenetic testing and dosing71: unavailability of testing at many medical centers, which leads to outsourcing of tests and a long turnaround time for results; lack of reimbursement for testing leading to large out-of-pocket expenses for patients (estimates run from $200 to $500); lack of support of genetic testing by professional organizations; and lack of clinician acceptance and knowledge of interpreting and applying test results. The 9th edition of the Antithrombotic Therapy and Prevention of Thrombosis: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines recommends against the routine use of pharmacogenetic testing when initiating a patient on warfarin.65 Genetic testing, if utilized, should be used along with patient characteristics, clinical considerations, and continued INR monitoring for optimal outcomes associated with warfarin use.
Dabigatran and rivaroxaban are two novel oral anticoagulants recently approved in the United States. Although genetic variants have not been well studied regarding these medications, there are some potential genes that may influence a patient’s response to these medications. The CYP enzymes are not important to the metabolism of dabigatran, although the CYP 3A4/5 and CYP2J2 do play a role in the metabolism of rivaroxaban and may serve as a cause of genetic variability in patients using rivaroxaban. Additionally, both dabigatran and rivaroxaban are P-glycoprotein substrates, which is encoded by the ABCB1 gene where several SNPs have been identified.71 Thus, future studies may be directed at elucidating whether the ABCB1 genotype, the CYP enzymes, and/or polymorphisms at the drugs’ target action site may serve as potential sources of variability in the dosing of these agents.
Activated Partial Thromboplastin Time
Normal range: varies by manufacturer, generally between 22–38 sec; therapeutic range for heparin-treated patients is 1.5–2.5 times control aPTT
The activated partial thromboplastin time (aPTT) is used to screen for deficiencies and inhibitors of the intrinsic pathway (factors VIII, IX, XI, and XII) as well as factors in the final common pathway (factors II, V, and X). The aPTT is also commonly used as a surrogate assay to monitor UFH and DTIs. The aPTT, reported as a clotting time in seconds, is determined by adding an aPTT reagent and calcium to the patient’s blood sample. The reagent contains phospholipids from animal, human, vegetable, or recombinant sources, as well as surface activators including kaolin, silica, or elegiac acid.73 Normal ranges vary depending on the reagent/instrument combination used to perform the test, but they generally are between 25–35 seconds.
Factor and heparin sensitivity as well as the precision of the aPTT test depend both on the reagents and instrumentation. In addition, some aPTT reagents are formulated for increased sensitivity to lupus anticoagulants. Despite numerous attempts to standardize the aPTT, very little progress has been made. The difficulty in part may reflect differences in opinion as to the appropriate heparin sensitivity, the need to have lupus anticoagulant sensitivity for targeted patient populations, and suitable factor sensitivity to identify deficiencies associated with increased bleeding risk. Normal and therapeutic ranges must be established for each reagent instrument combination, and ranges should be verified with changes in lots of the same reagent. Laboratory errors may cause either prolongation or shortening of the aPTT; these may include inappropriate amount and/or concentration of anticoagulant in the collection tube, time between collection of the blood specimen and performing the assay, inappropriate collection site (i.e., through a venous catheter, which contains heparin), and/or inappropriate timing of blood collection.63
Causes of aPTT prolongation. In addition to reagent specific issues impacting on aPTT responsiveness, hereditary diseases, or other acquired causes may prolong aPTT test results. Causes of aPTT prolongation include
- Hereditary
- – Deficiency of factor VIII, IX, XI, XII, prekallikrein, or high molecular weight kininogen (HMWK) (PT is normal)
- – Deficiency of fibrinogen or factor II, V, or X (PT is also prolonged)
- – Deficiency of factor VIII, IX, XI, XII, prekallikrein, or high molecular weight kininogen (HMWK) (PT is normal)
It should be noted that aPTT reagents may respond differently if a patient has a single factor deficiency versus a multiple factor deficiency; patients with single factor deficiency have a more predictable aPTT prolongation compared to patients with multiple factor deficiencies.74
- Acquired
- – Lupus anticoagulant (PT usually normal)
- – Heparin (PT less affected than aPTT, PT may be normal)
- – Lepirudin, bivalirudin, or argatroban (PT usually also prolonged)
- – Liver dysfunction (PT affected earlier and more than aPTT) (Minicase 1)
- – Vitamin K deficiency (PT affected earlier and more than aPTT)
- – Warfarin (PT affected earlier and more than aPTT)
- – DIC (PT affected earlier and more than aPTT)
- – Specific factor inhibitors (PT normal except in the rare case of an inhibitor against fibrinogen, factor II, V, or X)
- – Decreased nutritional intake; malabsorption
- – Myeloproliferative disease
- – Lupus anticoagulant (PT usually normal)
Though used to detect clotting factor deficiencies, today, the aPTT is used primarily for monitoring therapeutic heparin therapy. The generally accepted therapeutic range of heparin is an aPTT ratio of 1.5–2.5 times control, though this has not been confirmed by randomized trials.75 Given the inter- and intrapatient variability that can result from aPTT reagents, alternative means of monitoring heparin therapy are being scrutinized. This 1.5–2.5 aPTT ratio corresponds to75
- A plasma heparin concentration of 0.2–0.4 units/mL by assay using the protamine titration method
- A plasma heparin concentration of 0.3–0.7 units/mL by assay using the inhibition of factor Xa
Unfractionated heparin should be given by continuous IV infusion or subcutaneous injection, with exact dosing dependent on the indication. The aPTT should be drawn at baseline, 4–6 hours after continuous IV heparin is begun, and 4–6 hours after each subsequent dosage adjustment, since this interval approximates the time to achieve steady-state levels of heparin. Institutions may have their own specific heparin dosing nomogram or base their nomogram off one used in clinical studies; studies have shown several benefits to using a heparin nomogram, including decreased time to therapeutic anticoagulation, decreased time to achieving target aPTT levels, and decreased number of recurrent VTE thromboembolism episodes.76-82 Nomograms also allow quick fine-tuning of anticoagulation by nurses without continuous physician input.
aPTT determinations obtained earlier than 6 hours, when a steady-state concentration of heparin has not been achieved, may be combined with heparin concentrations for dosage individualization using non-steady-state concentrations. This approach has been demonstrated to reduce the incidence of subtherapeutic aPTT ratios significantly during the first 24 hours of therapy.83,84 This finding is important because the recurrence rate of thromboembolic disease increased when aPTT values were not maintained above 1.5 times the patient’s baseline aPTT during the first 24 hours of treatment.85,86
Warfarin effect on aPTT. Although warfarin also elevates the aPTT, aPTT is not used to monitor warfarin therapy. Studies have shown a strong correlation between the increase in PT/INR and a corresponding increase in aPTT with warfarin therapy.87,88 Therefore, if warfarin is started in a patient receiving heparin, the clinician should expect some elevation in aPTT.
Heparin concentration measurements may provide a target plasma therapeutic range, especially in unusual coagulation situations such as pregnancy, where the reliability of clotting studies is questionable. In this setting, shorter than expected aPTT results in relation to heparin concentration measurements may be indicative of increased circulating levels of factor VIII and increased fibrinogen levels.89 Patients may have therapeutic heparin concentrations measured by whole blood protamine sulfate titration or by the plasma anti-Xa heparin assay. However, they may have aPTTs not significantly prolonged above baseline. This difference has been referred to as a dissociation between the aPTT and the heparin concentration.90 Many of these patients have very short pretreatment aPTT values.
Decreased aPTT levels. While most attention has been focused on causes of prolonged aPTT levels, there is growing evidence of adverse events associated with decreased aPTT levels, with frequencies ranging from 6% to 20% of all aPTT tests performed (not including errors related to inappropriate blood collection).91,92 Several reports have stated that a shortened aPTT may indicate a risk factor for hypercoagulability.93-99 Clotting factors of the intrinsic pathway as well as vWF levels and activity have been elevated in some patients presenting with decreased aPTT levels, which provides some evidence that patients with decreased aPTT levels are hypercoagulable.92 However, it should also be noted that shortened aPTT levels may be associated with other conditions such as acute bleeding, increased in-hospital mortality, impaired fasting glucose, or diabetes, hyperthyroidism, and myocardial infarction.73,100-104 There is no definitive answer whether a shortened aPTT is a cause, consequence, or just an association with these other conditions. To rule out whether a shortened aPTT is due to a laboratory error, such as inappropriate specimen collection, a repeat collection and repeat testing should be performed.91
Heparin alone has minimal anticoagulant effects; when it is combined with AT (normal range: 80% to 120%), the inhibitory action of AT on coagulation enzymes is magnified 1000-fold resulting in the inhibition of thrombus propagation. Patients who are AT deficient (<50%) may be difficult to anticoagulate, as seen with DIC (Minicase 3). The DIC syndrome is associated not only with obvious hemorrhage but also with occult diffuse thrombosis.
A Patient on Anticoagulants
BRENDA C., A 56-YEAR-OLD FEMALE with atrial fibrillation and hypertension, presented to the emergency department with signs and symptoms of a new DVT. A routine heparin regimen was started. Brenda C. had the following laboratory determinations performed 6 hours after initiation of heparin:
| LABORATORY RESULTS | NORMAL RESULTS | PRETREATMENT PATIENT RESULTS | POSTHEPARIN RESULTS |
| PT (sec) | 10–13 | 11.1 | 12.3 |
| INR | 1+0.1 | 0.98 | 1.18 |
| aPTT (sec) | 21–45 | 19 | 23 |
| ATIII (%) | 80–120 | 57 | 50 |
Question: What might account for this Brenda C.’s postheparin elevation in PT? Why was the aPTT not prolonged? What long-term anticoagulation strategies are potential options for Brenda C.?
Discussion: Brenda C. was started on a continuous IV heparin infusion with plans to convert to long-term warfarin, per the institution’s protocol. Subsequent postheparin laboratory determinations revealed a hypercoagulable state consistent with thromboembolic disease. Circulating procoagulants account for the patient’s low pretreatment aPTT. ATIII—the cofactor with which heparin binds to exert its anticoagulant effect—is depressed, making the patient hypercoagulable. Subsequent to the initiation of heparin, the ATIII concentration declined further. This drop reflected the binding of ATIII to heparin.
Despite a normal dosing protocol for heparin, Brenda C.’s initial aPTT value (obtained 6 hours after the loading dose and continuous maintenance infusion of heparin) remained low. One might be suspicious of pseudoheparin resistance or dissociation of the aPTT responsiveness after adequate treatment with heparin. To assess the likelihood of this situation, a heparin concentration measurement could be obtained (discussed later) to demonstrate adequate heparinization despite a subtherapeutic aPTT determination.
Prior to initiation of concomitant warfarin therapy, Brenda C.’s PT increased slightly to 12.3 seconds. Minimal prolongation of the PT may be expected from heparin’s influence. This degree of elevation is consistent with this observation. Brenda C.’s INR should be followed, with the desired endpoint of warfarin therapy being an INR of 2–3. After 3 months, Brenda C. should be reassessed to see if further warfarin therapy is needed; she may have a choice between staying on warfarin longterm due to the atrial fibrillation, or she could switch to either rivaroxaban or dabigatran.
Six weeks later, Brenda C. presents for her regular INR followup. Her INRs have remained fairly stable between 2–3 while on warfarin 5 mg PO daily. She reports that her physician prescribed amiodarone 200 mg PO daily for rhythm control 3 weeks ago. Today her INR is 6.2, but she has no signs or symptoms of bleeding or bruising.
Question: How should her INR results be interpreted? What should be done next? How could this be prevented in the future?
Discussion: Brenda C.’s INR is supratherapeutic likely due to a drug interaction between warfarin and amiodarone. The clinician should also assess whether Brenda C. has reduced her overall intake of vitamin K-rich foods or if she has taken any extra doses of warfarin since these two factors may also increase the INR value (Table 16-7). Brenda C.’s dose of warfarin should be held for a few days and then reduced appropriately due to concomitant longterm amiodarone therapy. Her INR levels should be closely monitored during this time and for awhile thereafter. In the future, she should be instructed to inform her healthcare provider of any medication changes as soon as possible. If she chooses to switch to either dabigatran or rivaroxaban in the future, this drug interaction is no longer clinically relevant.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


