Hematology and Immunology
HEMATOLOGY
Anatomy/Histology
What cells are derived from pluripotent hematopoietic stem cells (think—what are the components of a CBC with differential)?
- Proerythrocyte → Reticulocyte → Erythrocytes (RBCs)
- Lymphoid stem cell → Lymphoblast → Lymphocytes (T and B cells*)
*B cells can go on to become plasma cells
- Myeloid stem cell → Monoblast → Monocytes
- Myeloid stem cell → Megakaryoblast → Megakaryocyte → Platelets
- Myeloid stem cell → Myeloblast → Promyelocyte → Myelocyte → Metamyelocyte → Band cell → Neutrophils, Eosinophils, Basophils
**Cell types listed in bold are components of a CBC
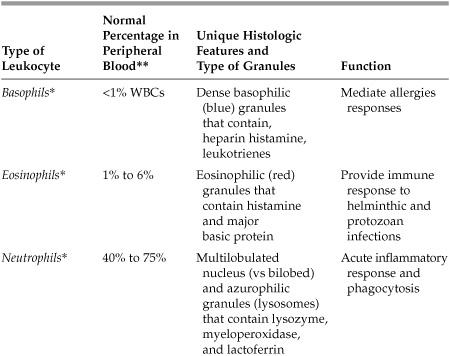
What are the various types of leukocytes (white blood cells) and what are their unique features?
See Table 5.1 on the following page.


Define erythrocytosis:
An increased number of RBCs (eg, as seen in polycythemia vera)
Define leukocytosis:
An increased number of WBCs (eg, as seen in infection or leukemia)
Define anisocytosis:
The presence of an increased amount of RBC size variation
Define poikilocytosis:
The presence of an increased amount of RBC shape variation
Define reticulocytosis:
An increased number of immature RBCs
Pathology—RBCs
What does the hemoglobin (Hb) on a CBC measure?
The concentration of hemoglobin in the blood (the normal range for men is 13-15 g/dL and for women is 12-15 g/dL).
What does the hematocrit (Hct) measure?
A percentage of the total volume of erythrocytes relative to the total blood in a sample. Typically, Hct = 3 × Hb. The normal range for men is 40% to 45% and for women is 35% to 45%.
What are the three major categories of anemia?
- Microcytic (MCV <80)
- Normocytic (MCV 80-100)
- Macrocytic (MCV >100)
What does the mean corpuscular volume (MCV) measure?
The average volume of red blood cells (RBCs). Since this is the average measurement it does not identify mixed cell populations, therefore a mixed anemia (microcytic and macrocytic may have a normal MCV).
What are common etiologies of microcytic anemia?
Iron deficiency anemia, thalassemia, lead poisoning, or anemia of chronic disease
*Iron deficiency is by far the most common
What are common etiologies of normocytic anemia?
Anemia of chronic disease hemolytic anemias, acute hemorrhage, aplastic anemias, renal failure
What are common etiologies of macrocytic anemia?
Vitamin B12 deficiency or folate deficiency (megaloblastic anemia), alcoholism, chronic liver disease, drugs that block DNA synthesis, significant reticulocytosis
Mechanistically, what causes anemia?
1. Decreased production of RBCs
a. Deficiency of nutrients (eg, iron) or proteins needed for hematopoiesis
b. Bone marrow failure
c. Decreased erythropoietin (typically secondary to renal failure)
2. Increased destruction or loss of RBCs
a. Hemolysis
b. Hemorrhage
To evaluate anemia of an unknown origin, what should you remember to order?
Reticulocyte count; this should be elevated with acute blood loss or hemolysis and low (<1%) with decreased RBC production.
What is the most common cause of iron depletion?
Chronic blood loss; seen often in menstruating women. In older patients and men, check stool for microscopic blood (ie, due to colorectal cancer).
What labs would you order to differentiate iron deficiency anemia from anemia of chronic disease?
See Table 5.2 below.
Table 5.2 Lab Values—Iron Deficiency Anemia versus Anemia of Chronic Disease

What causes α-thalassemia?
It is the result of mutations in α-globin genes leading to underproduction or absence of α-globin protein. There are four α-globin genes; severity of disease is a reflection of the number of genes involved.
- Missing 1 copy of the gene results in a silent carrier state; no anemia is present although patients may have slightly decreased MCV.
- Missing 2 copies of the gene leads to α-thalassemia trait, which typically manifests as mild microcytic hypochromic anemia and may be clinically mistaken for iron deficiency anemia.
What is hemoglobin H disease (“Hb H”)?
The form of α-thalassemia in which patient lacks three α-globin chains, leading to production of (β-tetramers. This results in mild to moderate microcytic hypochromic anemia with target cells and Heinz bodies, hepatosplenomegaly, and jaundice.
What is hemoglobin Barts (“Hb Barts”)?
All four α-globin genes are missing, leading to complete absence of α-globin chains and production of γ-tetramers and resulting in severe fetal anemia, hydrops fetalis, and often intrauterine fetal demise. In both Hb H and Hb Barts, the abnormal tetramers have a higher affinity for oxygen than normal hemoglobin resulting in impaired oxygen delivery to tissues.
*Bart = Babies die
What causes β-thalassemia minor and how does it present?
Underproduction of β chain (heterozygote). This is the milder form of β-thalassemia, with mild to moderate anemia.
What causes β-thalassemia major and how does it present?
Absence of β chain. Patients present early in childhood with severe anemia requiring repeated blood transfusions. Often patients have slowed growth and over time can develop sequelae of secondary hemochromatosis, including cardiac failure.
What are the major etiologies of hemolytic anemias?
- Autoimmune (idiopathic, drug related, or underlying disease)
- Nonimmune mediated (microangiopathic, hypersplenism, secondary to cardiac prosthesis, etc)
- RBC membrane defects—both acquired and congenital (hereditary spherocytosis, elliptocytosis, paroxysmal nocturnal hemoglobinuria [PNH], Wilson disease)
- Hemoglobinopathies (sickle cell disease, thalassemia, HbC)
- Enzyme defects (G6PD deficiency most commonly)
Mechanistically, what are the two ways that hemolysis occurs?
- Intravascular (as the name implies, RBCs destroyed within the blood vessels)—often more severe anemia; labs show hemoglobinemia, hemoglobinuria, and low haptoglobin (eg, microangiopathic, ABO incompatibility)
- Extravascular (aka within the reticuloendothelial system)—labs show elevated serum bilirubin and lactate dehydrogenase (LDH) (eg, RBC membrane defects)
What are the two categories of autoimmune hemolytic anemias?
- Warm agglutinin (IgG)—typically more chronic; seen in chronic disease (eg, SLE, CLL) and drugs; primarily extravascular hemolysis
- Cold agglutinin (IgM)—acute anemia; seen in infections such as mycoplasma and mononucleosis
What are schistocytes and how do they form?
Schistocytes are fragmented RBCs, resulting from mechanical/sheering damage to the RBCs, often seen in DIC or with mechanical heart valves.
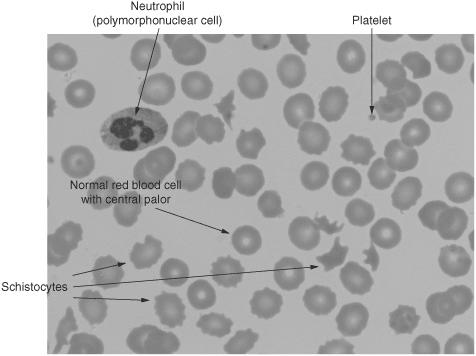
Figure 5.1 Schistocytes and bite cells among otherwise normal red blood cells, a single polymorphonuclear cell, and platelets. (Reproduced, with permission, from OHSU.)
Small round erythrocytes that have lost their central pallor and biconcave shape, formed secondary to a defective cytoskeletal protein. The sphere is the shape with the least surface tension but also the least flexible, resulting in frequent damage to the cell as it passes through the reticuloendothelial system (ie, the spleen).
What cytoskeletal proteins are defective in hereditary spherocytosis?
Spectrin and ankyrin
What test is used to confirm hereditary spherocytosis?
Osmotic fragility test
What is the definitive treatment for hereditary spherocytosis?
Splenectomy. Howell-Jolly bodies (basophilic nuclear remnants) are seen on blood smear after splenectomy.
What specific genetic mutation results in sickle cell anemia?
Single amino acid replacement of glutamine with valine on the hemoglobin β chain. Mutated hemoglobin is referred to as Hb S.
How does the mutation in sickle cell anemia actually lead to anemia?
The mutation in hemoglobin leads to decreased red blood cell deformability/elasticity. As the RBCs traverse the capillaries they do not have the normal elastic properties to distort as they pass through, instead the low oxygen tension promotes “sickling” of the cells. The repeated “sickling” of the cells damages the cell membrane, this damage in addition to the misshapen nature of the cells leads to increased destruction of RBCs in the spleen.
What are some clinical findings associated with sickle cell anemia?
Anemia, cholelithiasis, pain crisis, dactylitis (painful and swollen hands; and feet), and autosplenectomy
What are some other hemoglobinopathies?
- Hemoglobin C—similar clinical picture as HbS; most common in West Africa
- Hemoglobin E—range of illness from asymptomatic to severe; most common in Southeast Asia
What two microscopic findings are associated with glucose-6-phosphate dehydrogenase (G6PD) deficiency?
- Heinz bodies—membrane-bound precipitants of denatured hemoglobin secondary to oxidation of iron; can result in bite cells.
- Bite cells—partially devoured RBCs, where macrophages have taken a “bite” out, typically to remove a Heinz body.
How does disseminated intravascular coagulation (DIC) occur?
Coagulation sequence is activated; microthrombi form; platelets, fibrin, and coagulation factors are consumed; and fibrinolytic mechanisms begin
What are the common causes of DIC?
Sepsis, trauma, malignancy (particularly acute promyelocytic leukemia), obstetric complications (eg, preeclampsia), transfusions
What lab findings characterize DIC?
- High—prothrombin time (PT), partial thromboplastin time (PTT), fibrin split products (D-dimer)
- Low—platelet count
What is aplastic anemia?
Failure or destruction of pluripotent bone marrow precursor cells, resulting in pancytopenia
What WBC finding is associated with macrocytic/megaloblastic anemia?
Hypersegmented neutrophils seen in vitamin B12/folate deficiency
Pathology—Coagulation and Platelets
Which coagulation factor is deficient in hemophilia A?
Factor VIII
Which coagulation factor is deficient in hemophilia B?
Factor IX
What is the most common bleeding disorder?
von Willebrand disease
How does a deficiency in von Willebrand factor (vWF) lead to increased bleeding?
Directly results in impaired platelet adhesion and also decreased half-life of factor VIII.
It is stored within Weibel-Palade bodies within the vascular endothelial cells.
Which lab value measures the activity of the extrinsic pathway of the coagulation cascade?
Prothrombin time (PT): from this measurement a standardized number is calculated and reported as the INR (international normalized ratio).
Which lab value measures the activity of the intrinsic pathway?
Partial thromboplastin time (PTT)
What medication prolongs the PT/INR (with a normal PTT)?
Warfarin—the INR is typically monitored closely in patients on warfarin therapy.
What disease processes prolong the PT/INR?
Liver disease, vitamin K deficiency, DIC (early), Factor VII deficiency, lupus anticoagulant
What medication prolongs the PTT?
Heparin
What disease processes prolong the PTT (with a normal PT)?
Factor deficiencies (VII, IX, XI, XII), clotting factor inhibitors, von Willebrand disease, lupus anticoagulant
What is immune thrombocytic purpura (ITP)?
An autoimmune disorder where autoantibodies form against platelets
What population most often gets ITP?
Young women, 20 to 40 years old
What characterizes ITP?
Prolonged bleeding time with normal PT and PTT. The patient has pinpoint (petechial) hemorrhages, easy bruising, ecchymoses, low platelet count, but an increased number of megakaryocytes in the bone marrow.
What is thrombotic thrombocytopenic purpura (TTP)?
A condition with widespread formation of hyaline thrombi and consumption of platelets that leads to thrombocytopenia and microangiopathic hemolytic anemia
What might you see microscopically in TTP?
Schistocytes (fragmented red blood cells)
What is the pentad of symptoms seen in TTP?
Fever, thrombocytopenia, microangiopathic hemolytic anemia (MAHA), neurological changes, and renal failure
What causes Bernard-Soulier disease?
Mutation in Gplb resulting in a defect in platelet adhesion
What causes Glanzmann thrombasthenia?
Mutation in GpIIb/IIIa, again resulting in defective platelet aGGregation
*G for Glanzmann and aGGregation
What is the most common congenital thrombophilia?
Factor V Leiden—a mutation in Factor V (replacement of arginine with glutamine) conferring resistance to degradation by protein C and therefore leading to increased active factor V and a prothrombotic state. Other less common congenital thrombophilias include: prothrombin G20210A mutation, protein C deficiency, protein S deficiency, and antithrombin deficiency.
Give two examples of common acquired thrombophilias/hypercoagulable states:
- Malignancy
- Antiphospholipid antibodies (anti-cardiolipin antibodies and lupus anticoagulants)
IMMUNOLOGY
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


