Chapter 16 Hematology
Heparins




MOA (Mechanism of Action)
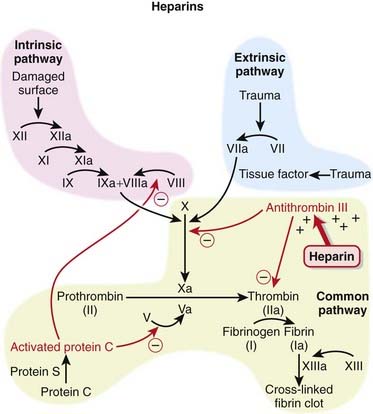
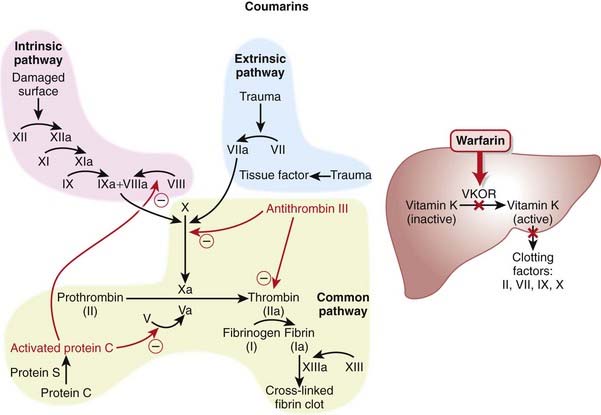
 The coagulation (clotting) system is composed of many proteins. Most of these proteins are procoagulants, which means they contribute to clotting. Some proteins are anticoagulants that serve to keep the coagulation system in balance.
The coagulation (clotting) system is composed of many proteins. Most of these proteins are procoagulants, which means they contribute to clotting. Some proteins are anticoagulants that serve to keep the coagulation system in balance. When a protein is activated, its name is followed by a small letter a. Each activated protein serves as an enzyme for the next protein downstream in the cascade (Figure 16-1).
When a protein is activated, its name is followed by a small letter a. Each activated protein serves as an enzyme for the next protein downstream in the cascade (Figure 16-1).


 To inhibit factor IIa, heparins must bind to both antithrombin III and factor IIa (dual binding not shown Figure 16-1).
To inhibit factor IIa, heparins must bind to both antithrombin III and factor IIa (dual binding not shown Figure 16-1).
Pharmacokinetics
Unfractionated Heparin
 Very complex pharmacokinetics exist because there is a large range in molecular weight of the molecules
Very complex pharmacokinetics exist because there is a large range in molecular weight of the molecules






Indications
 Treatment and prevention of inappropriate thrombosis
Treatment and prevention of inappropriate thrombosis










Important Notes
 To measure the effect of UFH, the aPTT test is used (compare this with prothrombin time [PT] or International Normalized Ratio [INR], which is used with warfarin). It is a measure of time to coagulation (in the laboratory) and is measured in seconds. The higher the number, the more strongly a patient is anticoagulated.
To measure the effect of UFH, the aPTT test is used (compare this with prothrombin time [PT] or International Normalized Ratio [INR], which is used with warfarin). It is a measure of time to coagulation (in the laboratory) and is measured in seconds. The higher the number, the more strongly a patient is anticoagulated.




Evidence

LMWH versus Warfarin for Treatment of Venous Thromboembolism
 A 2001 Cochrane review (seven studies, N = 1137 participants), updated in 2003, compared warfarin with LMWH for long-term treatment of VTE. There was no difference in the risk of recurrent VTE between warfarin and LMWH. There was a lower risk of bleeding with LMWH (odds ratio [OR] 0.38), and no difference in mortality rates between these two interventions was found.
A 2001 Cochrane review (seven studies, N = 1137 participants), updated in 2003, compared warfarin with LMWH for long-term treatment of VTE. There was no difference in the risk of recurrent VTE between warfarin and LMWH. There was a lower risk of bleeding with LMWH (odds ratio [OR] 0.38), and no difference in mortality rates between these two interventions was found.LMWH and Heparinoids versus UFH for Ischemic Stroke
 A 2008 Cochrane review (nine studies, N = 3137 patients) compared LMWH and heparinoids (danaparoid) with UFH in patients with acute, presumed or confirmed ischemic stroke. The odds of developing a deep vein thrombosis (DVT) were reduced with LMWH compared with UFH (OR 0.55); however, the incidence of key clinical outcomes such as PE, death, and hemorrhage (intracranial or extracranial) was too small to provide a reliable comparison.
A 2008 Cochrane review (nine studies, N = 3137 patients) compared LMWH and heparinoids (danaparoid) with UFH in patients with acute, presumed or confirmed ischemic stroke. The odds of developing a deep vein thrombosis (DVT) were reduced with LMWH compared with UFH (OR 0.55); however, the incidence of key clinical outcomes such as PE, death, and hemorrhage (intracranial or extracranial) was too small to provide a reliable comparison.


Direct Factor Xa Inhibitors

MOA (Mechanism of Action)
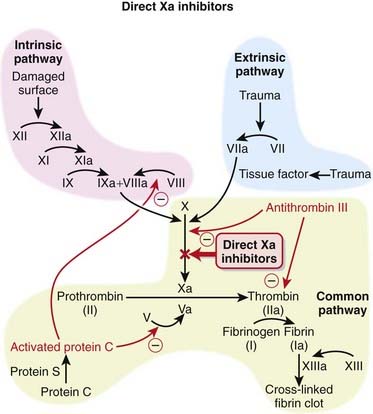
 The coagulation system is composed of many proteins: most of these proteins are procoagulants, which means they contribute to clotting. Some proteins are anticoagulants, which serve to keep the coagulation system in balance.
The coagulation system is composed of many proteins: most of these proteins are procoagulants, which means they contribute to clotting. Some proteins are anticoagulants, which serve to keep the coagulation system in balance. When a protein is activated, its name is given a small letter a at the end. Each activated protein serves as an enzyme for the next protein downstream in the cascade (Figure 16-2).
When a protein is activated, its name is given a small letter a at the end. Each activated protein serves as an enzyme for the next protein downstream in the cascade (Figure 16-2).




Pharmacokinetics
 Rivaroxaban, the first direct factor Xa inhibitor, is administered orally. It is rapidly absorbed, reaching peak plasma concentrations in 2 to 4 hours, and has an elimination half-life of 9 hours.
Rivaroxaban, the first direct factor Xa inhibitor, is administered orally. It is rapidly absorbed, reaching peak plasma concentrations in 2 to 4 hours, and has an elimination half-life of 9 hours.






Important Notes
 There are no monitoring requirements for rivaroxaban. This and the fact that it is an orally administered agent suggest that drugs in its class, and perhaps the direct thrombin inhibitors, will supplant warfarin as the drugs of choice among oral anticoagulants.
There are no monitoring requirements for rivaroxaban. This and the fact that it is an orally administered agent suggest that drugs in its class, and perhaps the direct thrombin inhibitors, will supplant warfarin as the drugs of choice among oral anticoagulants.Advanced
Drug Interactions
 CYP450 3A4 enzymes are involved in the metabolism of rivaroxaban; thus the potential exists for pharmacokinetic drug interactions with inhibitors or inducers of this isozyme. Rivaroxaban is also a P-glycoprotein (Pgp) substrate, and therefore its levels could also be affected by inhibitors or inducers of Pgp.
CYP450 3A4 enzymes are involved in the metabolism of rivaroxaban; thus the potential exists for pharmacokinetic drug interactions with inhibitors or inducers of this isozyme. Rivaroxaban is also a P-glycoprotein (Pgp) substrate, and therefore its levels could also be affected by inhibitors or inducers of Pgp.Evidence
Postsurgical Venous Thromboembolism Prophylaxis
 The RECORD trials were a series of double-blind randomized controlled trials that compared rivaroxaban with enoxaparin for the prophylactic treatment of VTE after total hip replacement (RECORD-1 and RECORD-2) or total knee replacement (RECORD-3 and RECORD-4). The trials were all relatively large, randomizing 2509 to 4541 patients between the two treatment groups. Rivaroxaban-treated patients had fewer events of VTE and all-cause deaths compared with enoxaparin in each of the four studies. The risk of bleeding was slightly higher with rivaroxaban than with enoxaparin.
The RECORD trials were a series of double-blind randomized controlled trials that compared rivaroxaban with enoxaparin for the prophylactic treatment of VTE after total hip replacement (RECORD-1 and RECORD-2) or total knee replacement (RECORD-3 and RECORD-4). The trials were all relatively large, randomizing 2509 to 4541 patients between the two treatment groups. Rivaroxaban-treated patients had fewer events of VTE and all-cause deaths compared with enoxaparin in each of the four studies. The risk of bleeding was slightly higher with rivaroxaban than with enoxaparin.FYI
 For a quick summary of the difference between two of the newer oral anticoagulants—direct factor Xa and direct thrombin inhibitors. Direct factor Xa inhibitors inhibit the formation of thrombin, whereas direct thrombin inhibitors allow thrombin to be formed but interfere with the actions of thrombin.
For a quick summary of the difference between two of the newer oral anticoagulants—direct factor Xa and direct thrombin inhibitors. Direct factor Xa inhibitors inhibit the formation of thrombin, whereas direct thrombin inhibitors allow thrombin to be formed but interfere with the actions of thrombin.Direct Thrombin Inhibitors




MOA (Mechanism of Action)
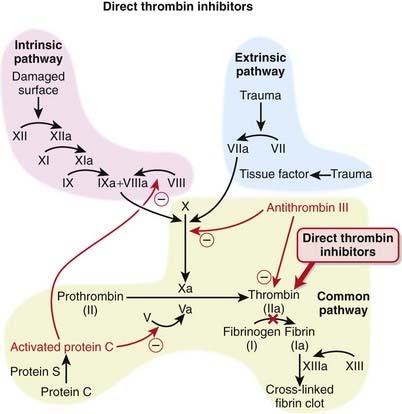
 The coagulation (clotting) system is a multistep cascade that eventually leads to the formation of fibrin and development of a clot.
The coagulation (clotting) system is a multistep cascade that eventually leads to the formation of fibrin and development of a clot. Thrombin (factor IIa) is an enzyme that catalyzes the final step in the coagulation cascade, the conversion of fibrinogen to fibrin (Figure 16-3).
Thrombin (factor IIa) is an enzyme that catalyzes the final step in the coagulation cascade, the conversion of fibrinogen to fibrin (Figure 16-3).













Important Notes
 All of the agents in this class are approved for prophylaxis of DVT and VTE after orthopedic surgery. Patients undergoing knee or hip replacement surgeries, in particular, are at high risk for developing a DVT or VTE. The risk is so high that these patients receive prophylactic anticoagulants for several days postsurgery.
All of the agents in this class are approved for prophylaxis of DVT and VTE after orthopedic surgery. Patients undergoing knee or hip replacement surgeries, in particular, are at high risk for developing a DVT or VTE. The risk is so high that these patients receive prophylactic anticoagulants for several days postsurgery.




Vitamin K Antagonists

MOA (Mechanism of Action)
 Vitamin K is key cofactor in the hepatic activation of four coagulation factors. The four vitamin K-dependent clotting factors are II, VII, IX, and X (Figure 16-4).
Vitamin K is key cofactor in the hepatic activation of four coagulation factors. The four vitamin K-dependent clotting factors are II, VII, IX, and X (Figure 16-4).





Indications
 Long-term (home) anticoagulation. Note that heparin would be used first for many of these conditions in the hospital and that warfarin would replace heparin on discharge.
Long-term (home) anticoagulation. Note that heparin would be used first for many of these conditions in the hospital and that warfarin would replace heparin on discharge.
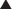




Important Notes
 Warfarin therapy is monitored using the International Normalized Ratio (INR), which is a standardized form of the PT test. Because different laboratories use different reagents to test the PT, every laboratory generates a slightly different result, so the INR corrects for this discrepancy among laboratories.
Warfarin therapy is monitored using the International Normalized Ratio (INR), which is a standardized form of the PT test. Because different laboratories use different reagents to test the PT, every laboratory generates a slightly different result, so the INR corrects for this discrepancy among laboratories.





Evidence
Warfarin versus LMWH for Treatment of Venous Thromboembolism
 A 2001 Cochrane review (seven studies, N = 1137 participants), updated in 2003, compared warfarin with LMWHs for long-term treatment of VTE. There was no difference in the risk of recurrent VTE between warfarin and LMWH. There was a lower risk of bleeding with LMWH (OR 0.38), and no difference in mortality rates between these two interventions.
A 2001 Cochrane review (seven studies, N = 1137 participants), updated in 2003, compared warfarin with LMWHs for long-term treatment of VTE. There was no difference in the risk of recurrent VTE between warfarin and LMWH. There was a lower risk of bleeding with LMWH (OR 0.38), and no difference in mortality rates between these two interventions.Warfarin versus Acetylsalicylic Acid for Atrial Fibrillation
 A 2007 Cochrane review (eight studies, N = 9598 participants) compared warfarin with acetylsalicylic acid (ASA) in atrial fibrillation patients who had not had a prior stroke or transient ischemic attack (TIA). Treatment with warfarin led to a lower risk of stroke (OR 0.68), ischemic stroke (OR 0.53), and systemic emboli (OR 0.48). The risk of intracranial hemorrhage was increased with warfarin (OR 1.98). All-cause mortality and vascular deaths were similar between groups, and disabling or fatal strokes and MI were almost reduced with oral anticoagulants, but this did not reach statistical significance.
A 2007 Cochrane review (eight studies, N = 9598 participants) compared warfarin with acetylsalicylic acid (ASA) in atrial fibrillation patients who had not had a prior stroke or transient ischemic attack (TIA). Treatment with warfarin led to a lower risk of stroke (OR 0.68), ischemic stroke (OR 0.53), and systemic emboli (OR 0.48). The risk of intracranial hemorrhage was increased with warfarin (OR 1.98). All-cause mortality and vascular deaths were similar between groups, and disabling or fatal strokes and MI were almost reduced with oral anticoagulants, but this did not reach statistical significance.

Salicylates





MOA (Mechanism of Action)
Salicylates
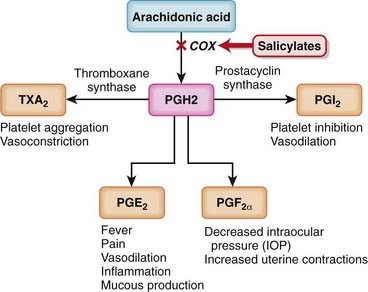
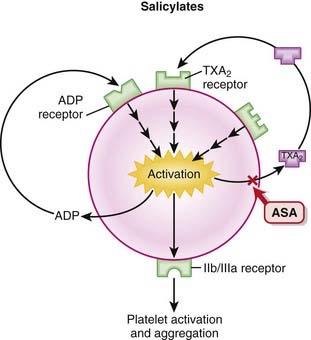
 Like nonsteroidal antiinflammatory drugs (NSAIDs), the salicylates work by inhibiting the actions of the cyclooxygenase (COX) enzyme (Figure 16-5).
Like nonsteroidal antiinflammatory drugs (NSAIDs), the salicylates work by inhibiting the actions of the cyclooxygenase (COX) enzyme (Figure 16-5). ASA works by irreversibly inhibiting COX-1, an enzyme that catalyzes the formation of cyclic endoperoxide, which in turn is then converted to the following:
ASA works by irreversibly inhibiting COX-1, an enzyme that catalyzes the formation of cyclic endoperoxide, which in turn is then converted to the following:
Aminosalicylates

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


