preload: this is governed by the ventricular end-diastolic volume, which in turn is related to ventricular filling pressure and therefore to venous return of blood to the heart,


 afterload: the systolic wall tension in the ventricle; this reflects the resistance to ventricular emptying within both the heart (during isovolumic contraction) and the peripheral circulation.
afterload: the systolic wall tension in the ventricle; this reflects the resistance to ventricular emptying within both the heart (during isovolumic contraction) and the peripheral circulation.The output from both right and left sides of the heart is normally balanced. In the healthy heart, cardiac output is regulated mainly by changes in heart rate and preload. Heart rate is modulated by the autonomic nervous system, with sympathetic nervous stimulation increasing heart rate and parasympathetic stimulation via the vagus nerve slowing the rate.
The relationship between preload and stroke volume (the amount of blood ejected from the ventricle during systole with each contraction) is shown in Figure 7.1. The degree of stretch of the ventricular muscle (preload) determines the force of cardiac contraction (the Frank–Starling phenomenon). The curve describing this relationship is governed by intrinsic myocardial contractility: thus, the curve is shifted upwards and to the left when contractility is augmented, for example by sympathetic nervous stimulation. In a healthy heart with normal myocardial contractility, the left ventricular filling pressure lies on the steep part of the curve, making stroke volume very sensitive to small changes in preload.
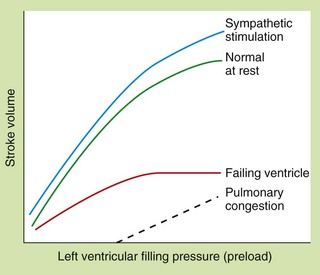
Fig. 7.1 The Frank–Starling relationship between preload (left ventricular filling pressure) and stroke volume in healthy and failing hearts.
In the severely failing heart, increases in filling pressure and heart rate are insufficient to restore cardiac output, and pulmonary congestion will occur.
The relationship between afterload and stroke volume is shown in Figure 7.2. Afterload is determined largely by peripheral resistance but also by the size of the ventricle. Enlargement of the left ventricular cavity (e.g. as a result of increased venous return or preload) increases wall tension, and the heart must generate greater pressure both to initiate and to maintain contraction. Preload and afterload are therefore interrelated. In the healthy ventricle a rise in afterload will cause a fall in stroke volume, but the consequent sympathetic stimulation will increase myocardial contractility and maintain stroke volume.
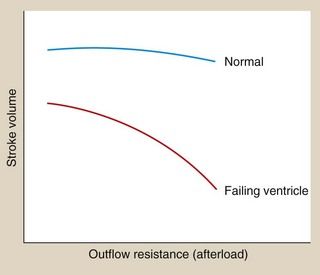
Fig. 7.2 The relationship between afterload (outflow resistance) and stroke volume in the presence of normal and reduced myocardial contractility.
Sympathetic stimulation maintains the stroke volume of the normal heart against an increasing afterload, but not in the failing heart.
Pathophysiology of heart failure
Heart failure is a syndrome that has several underlying causes (Box 7.1). Occasionally it arises suddenly such as after acute myocardial infarction or acute mitral regurgitation from rupture of the chordae tendineae. More commonly the onset is gradual from progressive loss of myocardial function or slow degenerative change in valve function.
Box 7.1 Causes of heart failure
Myocardial disease: cardiomyopathies, myocarditis
Congenital: atrial septal defect, ventricular septal defect, aortic coarctation
Infiltrative: amyloid, sarcoid, iron
Iatrogenic: β-adrenoceptor antagonists (high doses), anti-arrhythmics, calcium channel blockers, cytotoxics, alcohol, irradiation
The underlying problem in heart failure is reduced cardiac output and therefore low blood pressure, but the syndrome of heart failure arises largely from neurohumoral counter-regulation in response to low blood pressure and reduced renal perfusion. This is principally due to the sympathetic nervous system and the renin–angiotensin–aldosterone system (Fig. 7.3). The consequences of these compensatory mechanisms are vasoconstriction of both arteries and veins and excessive salt and water retention by the kidneys. Although these are the normal physiological responses to reduced blood pressure, in the setting of a failing heart they can create additional problems.
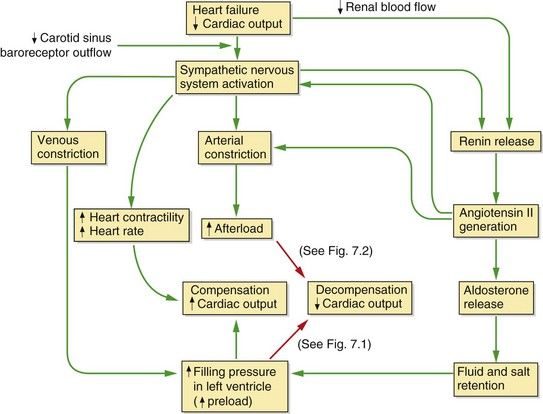
Fig. 7.3 Neurohumoral consequences of heart failure.
In the mildly impaired heart a fall in cardiac output results in a cascade of compensatory events (green arrows), including sympathetic stimulation of heart rate and contractility, constriction of arteries and veins, and activation of the renin–angiotensin–aldosterone system; overall these compensatory mechanisms restore cardiac output. If cardiac function is significantly impaired (red arrows), an increased preload cannot restore an adequate stroke volume (decompensation) (see Fig. 7.1) and the increased afterload will put additional strain on the failing heart and further decrease cardiac output (see Fig. 7.2). In chronic heart failure these effects are compounded by cardiac remodelling and downregulation of cardiac β1-adrenoceptors.
In the failing ventricle, the Frank–Starling curve is shifted downwards and to the right (failing-ventricle curve, Fig. 7.1) and the maximum achievable stroke volume is reduced. The curve is also flatter, indicating that stroke volume has become less responsive to changes in preload. As a result of activation of the compensatory mechanisms, salt and water retention expands plasma volume and venoconstriction enhances venous return to the heart. These factors increase the filling pressure of the left ventricle in an attempt to restore the resting stroke volume. Heart rate will also increase, which will raise cardiac output despite a lower stroke volume. If these responses are successful in restoring a normal resting cardiac output the heart failure is said to be compensated. However, the cardiac output may be unable to rise to meet the needs of the body during exertion.
Decompensation occurs when the combination of the increases in preload and heart rate fail to restore a normal resting cardiac output (Fig. 7.3). A persistent high level of symapathetic tone results in downregulation of β1-adrenoceptors and therefore less ability to maintain cardiac output. In most cases of heart failure, the impairment of function initially affects the left ventricle. As the central blood volume continues to increase in an attempt to raise the stroke volume, the hydrostatic pressure in the pulmonary veins will rise. When the hydrostatic pressure in the pulmonary circulation exceeds the plasma colloid osmotic (oncotic) pressure that holds fluid in the blood vessel, fluid leaves the capillaries into the interstitium of the alveoli and then into the alveolar spaces, producing pulmonary oedema (Fig. 7.2). Eventually, the raised pulmonary vascular pressure leads to right heart failure (producing biventricular failure, or congestive cardiac failure), and oedema develops in the peripheral and splanchnic tissues.
Peripheral arterial resistance (afterload) will also rise as a result of the compensatory mechanisms (Fig. 7.2). The failing ventricle cannot meet this with an increase in myocardial contractility so stroke volume will fall (Fig. 7.2) with further cardiac decompensation.
Heart failure arising from myocyte loss (such as occurs with myocardial infarction or cardiomyopathies) leads to adaptive changes in the surviving cells and extracellular matrix, known as remodelling. Remodelling is driven by several factors including local effects of catecholamines, angiotensin II, aldosterone and pro-inflammatory cytokines. This eventually produces a more globular, dysfunctional left ventricle. This type of heart failure is characterized by a reduced ejection fraction (heart failure with reduced ejection fraction or systolic heart failure).
In aortic or mitral valve regurgitation, heart failure arises because the left ventricle must accommodate the normal forward stroke volume and also the regurgitant volume (the volume leaking back into the left ventricle or left atrium respectively). Eventually, the left ventricle cannot enlarge sufficiently to maintain an effective stroke volume.
Heart failure can also arise from impaired diastolic relaxation even when contractile function in systole is normal. Stroke volume is reduced, but ejection fraction is normal. This is known as heart failure with preserved ejection fraction or diastolic heart failure. If the left ventricle fails to relax adequately, it will not accommodate the venous return, leading to pulmonary venous congestion and a low cardiac output, activating the same compensatory neurohumoral responses. Heart failure with preserved ejection fraction characteristically occurs in older people in association with left ventricular hypertrophy, but it also contributes to heart failure in ischaemic left ventricular dysfunction (see Ch. 5).
Symptoms in heart failure are caused by a reduced cardiac output (‘forward failure’) or venous congestion (‘backward failure’). The most common complaints are breathlessness from increased pulmonary venous pressure, and fatigue resulting from the reduced cardiac output and impaired skeletal muscle perfusion. In response to the reduced perfusion, biochemical changes also occur in skeletal muscle, making it less efficient. Other symptoms, such as the discomfort of peripheral oedema and anorexia due to bowel congestion, are attributable to a high systemic venous pressure. Increased stimulation of β-adrenoceptors in the heart can lead to life-threatening ventricular arrhythmias.
Acute left ventricular failure
Acute left ventricular failure usually results from a sudden inability of the heart to maintain an adequate cardiac output and blood pressure. It can follow acute myocardial infarction, acute mitral or aortic valvular regurgitation, or arise from the onset of a brady- or tachyarrhythmia if there is pre-existing poor left ventricular function. The sudden fall in cardiac output leads to reflex arterial and venous constriction (Fig. 7.3). There is a rapid rise in filling pressure of the left ventricle as a result of increased venous return. If the heart is unable to expel the extra blood, the hydrostatic pressure in the pulmonary veins rises until it exceeds the plasma oncotic pressure and produces acute pulmonary oedema. The principal symptom is breathlessness, usually at rest with orthopnoea.
Cardiogenic shock
The syndrome of cardiogenic shock arises when the systolic function of the left ventricle is suddenly impaired to such a degree that there is insufficient blood flow to meet resting metabolic requirements of the tissues. This definition excludes shock caused by hypovolaemia. The clinical hallmarks are a low systolic blood pressure (usually <90 mmHg), with a reduced cardiac output and an elevated left ventricular filling pressure. Cardiogenic shock can follow acute myocardial infarction, and in this situation usually indicates loss of at least 40% of the left ventricular myocardium. Other mechanical disturbances, such as acute mitral regurgitation or ventricular septal rupture, can produce cardiogenic shock in association with a lesser degree of myocardial damage. Less commonly, the syndrome is associated with right ventricular infarction. The mortality of cardiogenic shock, even with intensive treatment, is in excess of 70%.
Chronic heart failure
Myocardial damage from ischaemic heart disease is the most common cause of chronic heart failure, but potentially correctable causes such as valvular lesions, as well as treatable exacerbating factors such as anaemia or arrhythmias, may be identified. In most people with heart failure there are signs of both right and left ventricular failure (biventricular or congestive heart failure). Chronic heart failure is not a trivial complaint. People with left ventricular systolic dysfunction who have symptoms only on exertion have a 2-year mortality risk of about 20%, whereas the 1-year mortality is 80% if there are symptoms at rest. In heart failure with reduced ejection fraction the degree of left ventricular dysfunction is a guide to prognosis. Death is from either progressive heart failure or ventricular arrhythmias.
Positive inotropic drugs in the treatment of heart failure
Myocardial contractility can be improved by increasing the availability of free intracellular Ca2+ to interact with contractile proteins, or by increasing the sensitivity of the myofibrils to Ca2+. Only drugs that increase myocardial intracellular Ca2+ are established in clinical use; they work by one of two distinct mechanisms:


An additional advantage of the positive inotropic drugs that increase myocardial cAMP is their ability to enhance the reuptake of Ca2+ by the sarcoplasmic reticulum in diastole. This improves diastolic relaxation in addition to augmenting systolic contractility.
Digitalis glycosides
Mechanism of action and effects
Effect on myocardial contractility: Digitalis glycosides are compounds with a steroid nucleus that were originally isolated from a species of foxglove (Digitalis purpura). Digoxin is the drug that is most widely used in clinical practice. Digoxin binds to the energy-dependent Na+ pump (Na+/K+-ATPase) in the myocyte membrane. This pump establishes and maintains the Na+ and K+ gradients across the cell (Fig. 7.4), producing low intracellular Na+ and high intracellular K+ concentrations. A separate passive transmembrane exchange of Na+ and Ca2+ occurs down their concentration gradients, with Na+ entering the cell while Ca2+ is translocated out. The rate of this exchange is dependent on the intracellular Na+ concentration. Digoxin partially inhibits the Na+/K+-ATPase, which increases the intracellular Na+ concentration. This lowers the concentration gradient for Na+ across the cell membrane, which in turn reduces the Na+/Ca2+ exchange so that Ca2+ is retained in the cell. The excess intracellular Ca2+ is stored in the sarcoplasmic reticulum during diastole and released during cell membrane excitation, leading to enhanced myocardial contraction.
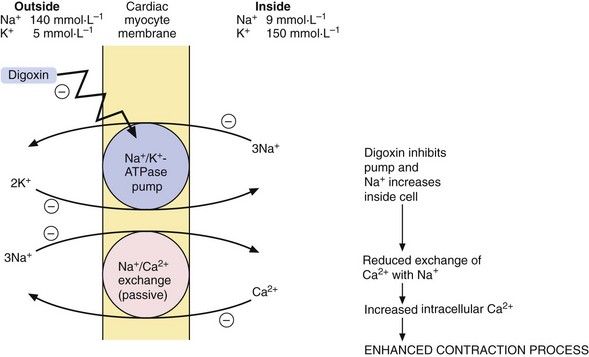
Effects on cardiac action potential and intracardiac conduction: Digoxin can be arrhythmogenic, but also has actions that are useful for treating certain arrhythmias.
Direct actions of digoxin on the heart can provoke arrhythmias by increasing myocardial cell excitability (Ch. 8), as follows.
 Reduction of the resting membrane potential. The cell membrane Na+/K+-ATPase pump extrudes three Na+ out of the cell for every two K+ that enter, which increases the negative intracellular electrical potential and hyperpolarises the cell, making it less excitable (see Ch. 8). Inhibition of this membrane pump by digoxin leads to the cell membrane potential becoming less negative and closer to the threshold potential for depolarisation. Arrhythmias are therefore more readily initiated.
Reduction of the resting membrane potential. The cell membrane Na+/K+-ATPase pump extrudes three Na+ out of the cell for every two K+ that enter, which increases the negative intracellular electrical potential and hyperpolarises the cell, making it less excitable (see Ch. 8). Inhibition of this membrane pump by digoxin leads to the cell membrane potential becoming less negative and closer to the threshold potential for depolarisation. Arrhythmias are therefore more readily initiated. Triggering of spontaneous release of Ca2+ from the sarcoplasmic reticulum. This leads to transient depolarisation of the cell immediately following an action potential (‘after-potentials’), which can initiate arrhythmias (Ch. 8).
Triggering of spontaneous release of Ca2+ from the sarcoplasmic reticulum. This leads to transient depolarisation of the cell immediately following an action potential (‘after-potentials’), which can initiate arrhythmias (Ch. 8).Digoxin also has useful indirect actions on the heart that arise from stimulation of the central vagal nucleus and enable it to be used for treating arrhythmias (Ch. 8). The vagal effects on the heart are:

 increased refractory period of the atrioventricular node, which slows impulse transmission to the ventricles and is useful in the management of the fast ventricular rates that result from atrial flutter and fibrillation (Ch. 8).
increased refractory period of the atrioventricular node, which slows impulse transmission to the ventricles and is useful in the management of the fast ventricular rates that result from atrial flutter and fibrillation (Ch. 8).Pharmacokinetics
Digoxin is well absorbed from the gut and the kidney is the main route of elimination, by filtration at the glomerulus and by active tubular secretion. The half-life of digoxin is very long (about 1.5 days) and is increased if renal function is impaired. The dose must be reduced in the presence of renal impairment to avoid toxicity (see below). If an early onset of action is required, loading doses should be given over the first 24 or 36 h (Ch. 2). If a rapid response is essential, digoxin can be given by slow intravenous injection.
Unwanted effects
Digitalis glycosides have a narrow therapeutic index, and toxicity is mostly dose-related.
 Gastrointestinal disturbances: anorexia, nausea and vomiting (largely a central effect at the chemoreceptor trigger zone; Ch. 32), and diarrhoea.
Gastrointestinal disturbances: anorexia, nausea and vomiting (largely a central effect at the chemoreceptor trigger zone; Ch. 32), and diarrhoea. Neurological disturbances: fatigue, malaise, confusion, vertigo, coloured vision (especially yellow halos around lights, possibly from inhibition of Na+/K+-ATPase in the cones of the retina).
Neurological disturbances: fatigue, malaise, confusion, vertigo, coloured vision (especially yellow halos around lights, possibly from inhibition of Na+/K+-ATPase in the cones of the retina). Distinctive changes on the ECG: this includes non-specific T-wave changes and sagging of the S–T segment with an upright T-wave (‘reverse tick’) often referred to as ‘digoxin effect’. These ECG effects do not indicate toxicity, but can be mistaken for myocardial ischaemia.
Distinctive changes on the ECG: this includes non-specific T-wave changes and sagging of the S–T segment with an upright T-wave (‘reverse tick’) often referred to as ‘digoxin effect’. These ECG effects do not indicate toxicity, but can be mistaken for myocardial ischaemia. Consequences of intracellular Ca2+ overload: increased excitability of the atrioventricular node and Purkinje fibres produces atrial or nodal ectopic beats, atrial or nodal tachycardia, ventricular ectopic beats or (less commonly) ventricular tachycardia.
Consequences of intracellular Ca2+ overload: increased excitability of the atrioventricular node and Purkinje fibres produces atrial or nodal ectopic beats, atrial or nodal tachycardia, ventricular ectopic beats or (less commonly) ventricular tachycardia. Consequences of increased vagal activity: excessive atrioventricular nodal block (‘heart block’) can occur. When associated with increased atrial excitability, this produces atrial tachycardia with atrioventricular nodal block, a rhythm characteristic of digitalis toxicity.
Consequences of increased vagal activity: excessive atrioventricular nodal block (‘heart block’) can occur. When associated with increased atrial excitability, this produces atrial tachycardia with atrioventricular nodal block, a rhythm characteristic of digitalis toxicity.Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


