When we discussed what epidemiologists do in Chapter 1, we touched on some of the different types of study that we use to collect the information we need to answer questions about health. In Chapter 3 we looked at the data systems and descriptive studies that provide the ‘bread-and-butter’ information of public health; in this chapter, we will look at the analytic studies that are our main tools for identifying the causes of disease and evaluating health interventions. Unlike descriptive epidemiology, analytic studies involve planned comparisons between people with and without disease, or between people with and without exposures thought to cause disease. They try to answer the questions ‘Why do some people develop disease?’ and ‘How strong is the association between exposure and outcome?’. This group of studies includes the intervention and cohort studies that you met briefly in Chapter 1, as well as case–control studies. Together, descriptive and analytic epidemiology provide information for all stages of health planning, from the identification of problems and their causes to the design, funding and implementation of public health solutions and the evaluation of whether they really work and are cost-effective in practice.
As we discussed in Chapter 1, people talk about many different types of epidemiology, but ultimately almost all epidemiology comes back to the same fundamental principles; the only things that differ are the health condition of interest and the factors that might influence that condition. When we discuss the various study designs in this chapter we will do so mainly in the context of looking for the ‘exposures’ that cause ‘disease’. However, the approaches that we will discuss are generic and are equally applicable to studies:
of treatment, prognosis and patient outcomes (e.g. survival, improved physical function, or quality of life) in clinical medicine, dentistry, nursing or any of the allied health professions;
of the effects of our occupation or our socioeconomic and physical environment on health;
aiming to identify factors that influence health behaviours such as smoking, alcohol consumption, or whether parents choose to have their children vaccinated;
evaluating programs that attempt to change behaviours in order to improve health outcomes;
evaluating the effects of changes in health practice or policy …
And the list could go on. Likewise, the range of exposures or study factors that might influence health – for good or bad – is incredibly broad. The ‘exposure’ we are interested in could be an environmental factor such as an infectious agent, radiation, or some chemical, it could be a behavioural factor like smoking or drinking habits, an intrinsic characteristic of the individual such as sex, age, skin colour, or an underlying genetic factor. Furthermore, while most of these are personal exposures that affect us at the individual level, epidemiology is expanding and social epidemiology encompasses the additional influences of the broader social environment. At another level, lifecourse epidemiology attempts to integrate exposures over an individual’s lifetime. While different questions place different demands on the specifics of data collection, all can be addressed via the same suite of research designs, although different designs will be more or less appropriate in different situations.
It is important to bear in mind that the study designs we discuss have their strengths, but they also have limitations, and we will touch briefly on these as we go. We will come back to pick up on some of these limitations in more detail when we talk about bias and confounding in Chapters 7 and 8 and when we look at how to report, read and interpret the results of epidemiological studies in Chapter 9. But first let us consider if there is an ideal study that would give us a completely unbiased picture of the effect of an exposure on an outcome.
Ignoring ethical and practical issues for a minute, what do you think would be the best way to determine whether factor A caused outcome B?
The ideal study
If a laboratory scientist wanted to see whether something caused a particular effect they would set up an experiment. This would involve creating two identical test systems under identical conditions, adding the particular factor of interest to one of them and then waiting to see what happened. Any differences between the outcomes in the two systems could then be fairly conclusively attributed to the presence of that factor (save for the play of chance, as discussed in Chapter 6). Unfortunately, life is not so straightforward when we are interested in human health.
One way to assess whether a factor affects health outcomes would be to compare the outcomes in a group of people exposed to the factor of interest to those among a group who were not exposed. If the outcomes differ, the challenge is then to decide whether it really was the exposure that caused the difference. For example, if we observe that people who exercise regularly (the ‘exposed’ group) are less likely to have depression than those who do not exercise (‘unexposed’), can we really be sure that physical activity is preventing depression?
What other reasons might explain the lower rate of depression among people who exercise?
In all likelihood, the two groups won’t differ only in their exposure status (level of physical activity), but they will also differ with respect to other factors that are correlated with the exposure. For example, people who exercise may have a more healthy diet, drink less alcohol, and be less likely to be overweight or to smoke than those who do not exercise. They will also be less likely to have chronic physical conditions that make exercise difficult. So how can we be sure it is not one of these other factors that led to the different rates of depression in the two groups? To rule out this possibility (a phenomenon known as confounding that we will come back to in Chapter 8) we would like our ‘exposed’ and ‘unexposed’ groups to be as similar as possible to each other in every respect except the exposure of interest, i.e. they need to be what is sometimes called exchangeable. In this situation, any difference in outcomes can only be due to the presence of the exposure in one group. The only way to be 100% sure that a particular exposure caused a specific outcome in a given individual or group of individuals would be to wind back the clock to see whether the same people would have experienced the same outcomes if they had lived lives identical in every way to their real lives, except they were now not exposed to the potential cause. If in this hypothetical situation the individual(s) did not develop the outcome, we could say with some certainty that the exposure did indeed cause the event. This imaginary parallel world which differs only from the real world with regard to the exposure of interest is often described as counterfactual (because it is contrary to fact) and, as epidemiologists, much of our effort is directed to designing studies that come as close to this hypothetical ideal as possible.
In this situation it is also possible that people who are depressed are less likely to exercise. This is often described as called reverse causality where it is the depression that causes inactivity rather than the other way around.
In practice, the closest that we can come to this is with what is known as a crossover trial in which the outcomes among a group of people exposed to the factor of interest are compared to the outcomes in the same group of people when they were unexposed. Although people cannot be exposed and unexposed at exactly the same time, as required by the counterfactual model, we hope the periods when they are exposed and unexposed are close enough together that nothing else changes. This type of study is, however, rarely possible in practice, so before we discuss it further we will take a step backwards to consider the epidemiological equivalent of a laboratory experiment – the intervention study or trial – more generally.
Intervention studies or trials
If there is good reason to believe that something might improve health then it is possible to conduct an intervention study where the investigator actively intervenes to change something to see what effect this has on disease occurrence. This is what James Lind did in his small, but classic study on scurvy in 1747 (Box 4.1). Such studies include clinical trials comparing two (or more) forms of treatment for patients with a disease, as well as preventive trials, in which the aim is to intervene to reduce individuals’ risk of developing disease in the first place. As with experiments in other sciences, the investigator controls who is exposed and who is not: for example, who is allocated to a new treatment regimen and who receives the old treatment, or who is enrolled in a ‘stop smoking’ campaign and who is not. Box 4.2 hints at the range of interventions that can be studied experimentally. In each study the investigators ‘intervened’ to change something in the hope that this would improve the future health of the participants. The participants in the ISIS trials were already sick (patients who had had a heart attack), and the intervention (treatment) was intended to increase their chances of surviving – aspirin and streptokinase were shown to be very effective. In contrast, the children in the polio vaccine trial were healthy and it was hoped that the vaccine would prevent them from becoming ill. Similarly, it was hoped that vitamin A supplementation would reduce childhood mortality but, in this example, the intervention was given to whole villages rather than individual children.
In 1747, James Lind conducted an experiment to test six different cures for scurvy. While at sea, he identified 12 patients with scurvy whose ‘cases were as similar as I could find them’ and prescribed a different treatment to each pair of patients. After a few days he found that the two patients fortunate enough to have been prescribed oranges and lemons were almost fully recovered whilst no improvement was seen in the other 10, who had been subjected to various regimens including seawater, gruel, cider and various elixirs. From this, Lind inferred that inclusion of citrus fruit in the diet of sailors would not only cure, but also prevent scurvy (Lind, 1753). Limes or lime juice thus became a part of the diet on ships, earning British sailors their nickname of ‘limeys’.

In the early 1950s, one of the largest epidemiological studies, and almost certainly the largest formal human ‘experiment’, was conducted in the USA. This was a field trial of polio vaccine in which over 400,000 school children were assigned to receive either the vaccine or a placebo (inactive) injection. The trial clearly demonstrated both the efficacy and the safety of the vaccine, which was then given to millions of children throughout the world (Francis et al., 1955). This has led to a major drop in the incidence of polio in both industrialised countries and also many developing countries which have now been declared polio-free by WHO.
The ISIS (International Studies of Infarct Survival) investigators recruited more than 100,000 patients around the world into a series of trials testing treatments to prevent early death after a suspected myocardial infarction (heart attack). These treatments included aspirin and streptokinase which were found to be highly effective in ISIS-2 (ISIS-2 Collaborative Group, 1988).
In the US Physicians’ Health Study (we have already met the British Doctors and the US Nurses’ Health studies), 22,000 physicians were randomly allocated to take aspirin, in an attempt to prevent cardiovascular disease, and/or β-carotene, in an attempt to prevent cancer (Hennekens and Eberlein, 1985). After 12 years of follow-up, rates of cancer were very similar in the β-carotene and placebo groups, and, while aspirin was shown to lower the rates of heart attack, so few of these very healthy doctors died that the trial could not determine whether aspirin saved lives from cardiovascular disease.
A randomised, controlled community trial involving almost 26,000 preschool children was conducted to evaluate the effectiveness of vitamin A supplementation to prevent childhood mortality in Indonesia (Sommer et al., 1986). In 229 villages, children aged 1–5 years were given two doses of vitamin A while children in the 221 control villages were not given vitamin A until after the study. Mortality among children in the control villages was 50% higher than that in the villages given vitamin A.
Randomised controlled trials (RCTs)
The best way to evaluate a new treatment is to identify a group of patients with the same condition and then randomly allocate them to either receive the treatment or to a control group that does not receive the treatment. A preventive trial differs only in that it involves people who are disease-free but thought to be at risk of developing disease. Random allocation (also called randomisation) of individuals to the study groups is the only way to ensure that all of the groups are as similar as possible (i.e. exchangeable) at the start of the study. It is important because if one group were in some way more ill (or less healthy) than the other at the start, this might make this group appear to have worse outcomes, even if the intervention really had no effect (another example of confounding). While we can look for and deal with some factors that differ between study groups in our analysis, there may also be other important factors that we either do not know about or cannot measure well. The real strength of randomisation is that, on average, it will also balance these other unknown or poorly measured factors across the groups. It is because of this aspect of RCTs – the close similarity of the groups in all respects other than the intervention – that they are generally considered to give the best evidence of all epidemiological studies.
There is an important distinction between ‘random selection’ where we select people at random to be in our study but we do not control whether or not they are exposed (unless it is a RCT), and ‘randomisation’ where we do control exposure by randomly allocating people to the exposed and non-exposed groups in an intervention study.
Randomisation does not always work. Equality of the groups at baseline is highly dependent on group size. Even if participants are allocated to groups at random, if the groups are small it is unlikely that all of the factors that could affect the outcome will be evenly distributed across the groups.

The control or comparison group is essential so that outcomes in the treated group can be compared with the outcomes among similar people who have not been treated. Sometimes the patients in the control group receive no treatment but, preferably, they are given a placebo (something that resembles the real treatment but is not active). And if an acceptable standard treatment is available the control group must be given this – it would be unethical to withhold it – and this is compared with the new experimental treatment. Figure 4.1 shows the design features of a simple RCT.
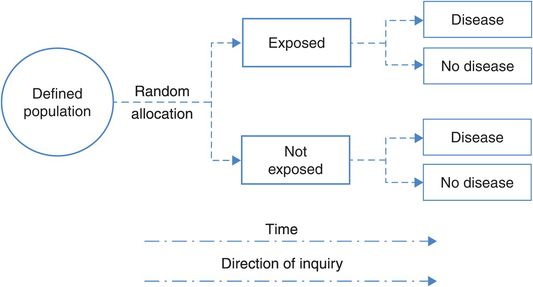
The design of a randomised controlled trial.
Ideally, both the trial investigators and the participants should be unaware of whether the participant is in the active intervention or placebo group, creating a ‘double-blind’ or ‘masked’ study. If only the patient is unaware of their allocation, it is a single-blind study. Blinding is important because knowledge of the treatment might affect both the participant’s response and an observer’s measurement of outcome. If the participant knows they have been given the new treatment they might feel better simply because they believe it will do them some good (a placebo effect) and, likewise, an observer might be more likely to report signs of improvement in someone if they know they received the active treatment. In some situations (e.g. comparing medical treatment with surgery) there may be no feasible way of blinding patients and study personnel to the differences in treatments. Minimising measurement bias in this situation may be best accomplished by bringing in an independent ‘blinded’ observer whose only involvement is to assess the outcome measure. Blinding of outcome measurements obviously becomes more crucial as the measurement becomes more subjective. When the outcome is objective and less dependent on interpretation, as in a biochemical parameter or death, blinding is less important.
Apart from participants and trial investigators, there are many others (e.g. health care providers, data collectors, outcome assessors, data analysts) involved in the conduct of a trial who can introduce bias through their knowledge of treatment allocation. For this reason there is a growing tendency to abandon the terms single- and double-blind in favour of a transparent reporting of the blinding status of each group involved in the trial.
The other crucial feature of an RCT is good follow-up. It is important to know what has happened to all of the participants in the study because if many people are ‘lost to follow-up’ such that we don’t know if they experienced the health outcome of interest, then the results of the study may be biased. This is a form of selection bias and we will discuss it further in Chapter 7.
Crossover trials
The trials we discussed above and shown in Figure 4.1 are what are known as parallel group trials, in which individuals are randomly allocated to one of the two groups, which are then followed in parallel. In a crossover design, the participants serve as their own controls and thus, as described above, we come closer to achieving the counterfactual ideal where we observe the same people twice under conditions that are identical except with respect to the exposure of interest. For example, in a simple two-period crossover study to assess the efficacy of an intervention we would randomly assign each participant to either the intervention or the control (I or C) for a specified period of time and then the alternative for a similar period of time. Thus, approximately half of the participants would receive the interventions in the sequence I–C and the other half in the sequence C–I, reducing the impact of any factors that might change between the first and second period.
One of the biggest advantages of this design is that it removes much of the variability that is inherent when we compare different groups of people and that can never be completely eliminated by randomisation. In particular, it ensures the groups are truly exchangeable from a genetic perspective and in terms of other factors that do not change over time. As a result, crossover trials can produce statistically and clinically valid results with fewer participants than would be required with a parallel design. However, it is important to remember that while we are comparing the participants to themselves, time has moved on and so it is impossible to be completely sure that nothing else has changed. As a minimum it is likely that the weather will have changed and this may affect various aspects of behaviour. Also, not all interventions are suitable for assessment in this way. We can only use crossover trials to assess the effects of factors that have a rapid effect and where the effect wanes rapidly when the exposure is removed. If the effects of the intervention during the first period are likely to carry over into the second period then this design is clearly inappropriate, as is also true for assessing long-term benefits and harms.
n-of-1 trials
A variant of the crossover trial is the single patient trial, often called an n-of-1 trial. Here, an individual patient receives the experimental and control treatments in random order on multiple occasions, with specific outcomes being monitored throughout the trial period. Ideally both the patient and the treating doctor are blinded to the treatment being received and the trial usually ends when it becomes clear that there are (or are not) important differences between the treatments. As with the crossover trial, a strength of these studies is that by comparing the same person on and off treatment we come closer to achieving true exchangeability. Again, we have the problem that we are comparing the active treatment and control at different points in time but, by giving them multiple times in a random order, we hope to minimise any bias that might result from this. Although the results of n-of-1 trials are not generalisable to the same extent as those of typical RCTs, they do provide a good guide to individual clinical decisions.
“n” is often used to denote the sample size in a study; an n-of-1 study is thus a study where n = 1 person.
Cluster randomised controlled trials
For many public health and practice/policy interventions it is impossible to intervene at an individual level so the answer is a cluster randomised controlled trial where groups or clusters of people are randomised together. An example is a study that evaluated a ‘Comprehensive Health Assessment Program’ (CHAP) to enhance interaction between adults with intellectual disability, their carers and general practitioners. To avoid contamination whereby participants randomised to the control arm were inadvertently exposed to the intervention via their housemates, individuals living in the same house or who saw the same GP were randomised together as a ‘cluster’. In this study the clusters were of very different sizes so they were matched to another cluster of similar size and characteristics and one of each pair was allocated at random to the intervention and one to the control arm. The study showed increased detection of health problems and increased screening rates in the intervention group (Lennox et al., 2007).
Community trials
Community trials are cluster trials in which the intervention is implemented at the community level. They are generally conducted when it would be impossible to offer (or evaluate) the intervention at the individual level. An example is the studies of water fluoridation and dental health conducted in various countries. When investigators wanted to study the effects of adding fluoride to the water supply on dental health it was clearly impossible to add fluoride to some people’s water and not to others’, so whole towns were allocated to receive fluoride in their water or not. The controlled trial of water fluoridation which gave the most striking results was carried out in the towns of Newburgh and Kingston in New York State, USA. After 10 years of fluoridation, the DMF (decayed, missing or filled teeth) score for Newburgh children aged 6–16 was 50% lower than that for children in the unfluoridated town of Kingston (Ast and Schlesinger, 1956). The assumption underlying this result was that, apart from the water, there was no other major difference between the towns that could explain the effect (i.e., there was no confounding). (Note that, although this and other studies clearly showed the benefits of low levels of fluoride on dental health, continuing controversy about the possible adverse effects of fluoride on other organs in the body, particularly the bones, has meant that universal fluoridation of water supplies has not occurred.) Because only two towns were included in this study, in practice it is little different from a non-randomised comparison (see below). Other cluster designs involve larger numbers of groups, so that the random allocation of multiple groups to intervention or no intervention gives more of the benefits of randomisation in terms of balancing out other factors across the groups (e.g. the vitamin A study, Box 4.2).
Non-randomised designs
The fact that a study is described as a trial or clinical trial does not necessarily mean that it is a randomised controlled trial. While RCTs remain the gold standard for initial evaluation of new clinical and public health interventions, the effectiveness of these interventions in practice can often only be determined from ‘before and after’ type comparisons in whole communities or populations. You will see examples of this throughout the book, and particularly when we discuss prevention in Chapter 14 and screening in Chapter 15. Probably the most common non-randomised design in the health setting is one that uses ‘historical controls’ where health outcomes following the introduction of a new treatment or preventive measure are compared to the outcomes experienced by the same population before the change in practice (also sometimes called a pre–post study). For example, in many countries, mortality rates from road traffic accidents fell dramatically after the introduction of legislation requiring drivers to wear seat belts. Similarly, patient survival rates might be compared before and after the introduction of a new surgical technique.
More pre–post study designs: if measures are obtained at multiple time points before and after the intervention this may be described as an interrupted time series, while if multiple groups are involved it is sometimes called a multiple baseline design.
The main problem with this design is that it assumes that the only (or most important) thing that has changed is the new legislation or the type of surgery, and that may not be the case. Another variation on the cluster trial is the ‘stepped wedge’ design, where the intervention is sequentially introduced to all of the study groups. We will not discuss these designs further here, but see Sanson-Fisher et al. (2014) for further information.
Observational studies
Although the ideal way to study whether something is causally related to the occurrence of disease is through an experiment or intervention study, this would often be unethical (you cannot deliberately expose someone to something thought to be harmful) or impractical. As a result, epidemiology is rarely an experimental science. Most of the time an epidemiologist will just go out (after a lot of thoughtful planning) and measure the rate of occurrence of a disease or other health outcome, or will compare patterns of exposure and disease to identify particular exposures or risk factors associated with that disease. This is purely an observational role: the researcher does not intervene in any way. They leave nature to take its course, and record what happens, or what happened in the past. These are commonly described as observational studies.
Cohort studies
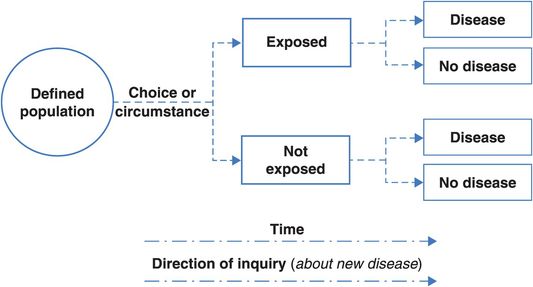
The next best thing to a randomised trial is a cohort study (sometimes described as a prospective or longitudinal study). Like a trial, we follow people forwards (prospectively) over time to see what happens to them and, again like a trial, the cohort might be a group of initially healthy people whom we follow to measure the occurrence of disease or a group of patients whom we follow to study their disease outcomes, i.e. their prognosis. Figure 4.2 shows the design of a typical cohort study.
In Ancient Rome, a cohort was one of 10 divisions of a Roman military legion. It comprised young men of similar age from one region. In service its members were often injured or killed and they were not replaced. The cohort was then disbanded when the term of enlistment was over.
Compare Figures 4.1 and 4.2. How does a cohort study differ from an RCT?
The fundamental difference is that in a trial the investigator controls who is exposed to the factor of interest and who is not, ideally by assigning people to the different exposure groups at random, whereas in a cohort study participants are living the lives they have chosen, and the researcher has to discover and measure the exposures they have chosen for themselves.
Why is this a problem?
The challenge with this type of study is to disentangle the effects of the exposure that we are interested in from those of other personal characteristics or behaviours that are correlated with that exposure. In a randomised trial these other factors should be distributed evenly across the study groups, but this is unlikely to be the case in a cohort study where people who choose to smoke, for example, may also be more likely to drink alcohol or coffee, or to exercise less and so on, i.e. the groups are intrinsically less similar (exchangeable) than the arms of a randomised trial.
One classic example of a cohort study is the Framingham Heart Study (Dawber, 1980). It was started in 1948 at a time when heart disease had become the USA’s number one killer, and the principal aim was to identify biological and environmental factors that might be contributing to the rapid rise in cardiovascular death and disability. The epidemiological approach was quite novel at the time and it was designed to discover how and why those who developed heart disease differed from those who escaped it. The town of Framingham, Massachusetts was selected by the US Public Health Service as the study site, and 5209 healthy men and women between 30 and 60 years of age were enrolled and followed over time to see who developed disease and/or died. Framingham was appealing because it had a stable population and a single medical facility, suggesting that it would be relatively easy to carry out the follow-up. The study was expanded in 1971 when 5124 children (and their spouses) of the original cohort were recruited for a second study, the Offspring Study.
Before Framingham, the notion that scientists could identify, and individuals could modify, risk factors (a term coined by the authors of the study) tied to heart disease, stroke and other diseases was not part of standard medical practice. With over 50 years of data collected from residents of Framingham (and the publication of more than 1000 scientific papers), the Framingham researchers have identified major risk factors associated with heart disease, stroke and other diseases and created a revolution in preventive medicine. The study identified several risk factors associated with increased risks of heart disease including cigarette smoking (1960), high cholesterol levels and high blood pressure (1967), obesity and low levels of physical activity (1967). These are so commonly accepted today, both by health professionals and by the public, that it is difficult to imagine a time when we did not know their importance.
The Framingham Study is quite small by modern standards: the European Investigation into Cancer (EPIC) established in 1995 includes more than half a million individuals from 10 European countries (Bingham and Riboli, 2004) and the Million Women Study was initiated in the UK in 1996 (The Million Women Study Collaborative Group, 1999). The Framingham study therefore needed particularly long follow-up to accumulate enough endpoints to give robust results. A crucial trade-off is that the smaller size and the setting permitted regular detailed physical examination and other ‘hands-on’ investigations such as the recording of electrocardiograms, giving a rich array of high-quality exposure data that cannot be gathered on a very large scale.
Of all the observational designs, cohort studies generally provide the best information concerning the causes of disease and the most direct and intuitive measurements of the risk of developing disease. The participants must be free of the outcome of interest at the start of the follow-up, which makes it easier to be confident that the exposure preceded the outcome (i.e. to rule out the possibility of reverse causality). However, if there is a long pre-clinical phase before a disease is diagnosed, as may be the case for many types of cancer, the apparent exposure–disease sequence can still be wrong, and for this reason many cohort studies do not count cases of disease that occur in the first few years of follow-up. The other advantage of collecting exposure data before people develop disease is that measurement of exposure is not biased by knowledge of outcome status, i.e. it avoids recall bias. It is important to note, however, that if a cohort study has a very long follow-up period and exposure data were only collected at baseline then people may have changed their behaviours over the intervening years. For example, smokers may quit smoking or meat eaters may become vegetarian and it is also an unfortunate fact that many of us will gain weight as we get older. Depending on when the critical period of exposure occurs, this may mean that people are wrongly classified with regard to their exposure (e.g. past smokers as current smokers). This is a problem of misclassification. Many cohort studies, for example the US Nurses’ Health Studies (see Box 4.3), avoid this problem by re-contacting study participants every few years to collect updated exposure data. (We will discuss misclassification and recall bias further in Chapter 7.)

The British Doctors Study cohort was established in 1951 and followed for more than 50 years, although most of the original 40,701 participants are now dead. It has been of enormous value, particularly in relation to identifying the manifold health consequences of smoking. This is despite the fact that, compared with studies today, only limited exposure data were collected on a very short postal questionnaire mailed to the doctors at 10-year intervals since 1951 (Doll and Hill, 1964; Doll et al., 2004).
The US Nurses’ Health Study (www.nurseshealthstudy.org/) started in 1976 with 121,964 female nurses aged 30–55, and 5 years of funding. Since then, its focus has widened enormously from the oral contraceptive–breast cancer links for which it was first funded (Stampfer et al., 1988) to cover many exposures (including diet) and a multitude of outcomes. It has now accumulated more than 30 years of follow-up and is still going strong. It is very expensive to run, but the scientific and public health yield has been exceptional. The Nurses’ Health Study II began in 1989 with 117,000 nurses aged 25–42 (Rockhill et al., 1998) and in 2010 they started the Nurses’ Health Study III, an entirely web-based study targeting nurses aged 20–46 years in the USA and Canada.
ALSPAC (The Avon Longitudinal Study of Parents and Children, www.bristol.ac.uk/alspac/) was started in 1990 to determine ways in which an individual’s genes combine with environmental pressures to influence health and development. Comprehensive data have been collected on over 10,000 children and their parents, from early pregnancy until the present. Because the study is based in one geographical area of the UK, linkage to medical and educational records is relatively simple, and hands-on assessments of children and parents using local facilities allows good quality control (Golding et al., 2001).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


