Healthcare-Associated Infections Following Transfusion of Blood and Blood Products
Healthcare-Associated Infections Following Transfusion of Blood and Blood Products
Charles J. Schleupner
Eyad Cheikh
The transfusion of blood and blood products exposes recipient patients to both noninfectious and infectious adverse events (1). Approximately 15 million units of blood are collected and processed into 29 million products annually, with an average of 2.7 units administered per recipient (2). In a recent review of noninfectious early and late adverse events after transfusion, mortality rate estimates are one per million to 8 million transfused components, much lower than prior estimates (0.0001-0.00001%; 3). Infections account for 8% to 17% of these deaths. The remainder of deaths are related to early or late, frequently immunologic, reactions, or to errors of processing or administration. The nonfatal adverse effects range from minor inconveniences to life-threatening emergencies; they may occur immediately during or within hours of the transfusion (early events) or may be delayed for weeks, months, or years (late events). The physician should always consider the risk of such ill occurrences in the decision to transfuse. The public desires absolute safety in a product regardless of cost (4,5); accordingly, there has been a marked increase of safety of transfused blood in the United States over the last 20 years (6,7, 8, 9, 10, 11and12). This chapter reviews the potential infections transmitted by blood and blood products (Table 67-1) and their prevention.
NONINFECTIOUS COMPLICATIONS
Noninfectious complications of blood transfusion have emerged as the most common cause of fatal transfusion reactions and have become the focus of hemovigilance by the National Healthcare Safety Network (NHSN; 13). The noninfectious complications of blood transfusion include acute transfusion reactions (within 24 hours) and delayed reactions (>24 hours after transfusion; 1,3,13). Acute reactions include acute hemolytic reactions, which may be immunologically or nonimmunologically mediated and account for the majority of deaths, and nonhemolytic reactions, which are usually febrile or allergic in nature and account for 19% to 25% of deaths resulting from transfusion (1,13). Other nonhemolytic reactions include volume overload, transfusion-related acute lung injury (TRALI), sepsis from bacterial contamination (vide infra), air embolus, hypertension associated with coadministration of an angiotensin-converting enzyme inhibitor, hypocalcemia, and hypothermia (1,3,13). Delayed reactions include hemolysis due to anamnestic antibody responses, alloimmunization to red blood cell (RBC) or human leukocyte (HLA) antigens, posttransfusion purpura, hemosiderosis, graft-versus-host disease (GVHD) in severely immunosuppressed hosts, and processing errors (1,3,13). Some of these noninfectious complications are obviated by routine measures. Most are not life threatening and can be medically managed when they occur. Macroaggregate leukocyte filters during transfusion may reduce some of these reactions (9,14). Leukocyte reduction (LR) before storage with third-generation filters is very important for reducing transfusion reactions associated with the presence of cytokines generated by leukocytes in the plasma phase of stored donor units (15,15a,16). Since implementation in Canada and European countries, LR has resulted in decreased transfusion-associated mortality, fever, transfusion-related immunomodulation (TRIM), and decreases in the incidence of TRALI and its associated mortality (17, 18, 19, 20and21). LR in the United States has resulted in similar benefits (3,13). Prestorage LR was advocated by the American Red Cross (ARC) in the summer of 1995 and by the summer of 2000, 95% of ARC donor centers had implemented this policy. In 2010, over 95% of ARC centers continue to use prestorage LR; many but not all non-ARC donation centers in the United States are doing likewise (personal communication, ARC).
INFECTIOUS COMPLICATIONS
It was not until the early 20th century that transfusion was made feasible for nonterminally ill patients. The new methods included anticoagulation techniques, classification of blood type isoagglutinins (22), and storage of donor blood. Previously, only direct donor-to-recipient transfusions were reluctantly and rarely performed because of the high frequency of often severe complications (23). With the advent of this technology, the infectious complications of blood transfusion became recognized. By World War I, potential donors with malaria, syphilis, and fever were excluded (24). The increased number of transfusions during World War II led to the recognition of posttransfusion hepatitis (PTH) (25). However, even before this description, the American Red Cross Donor Service had deferred all potential blood donors with a history of jaundice within 6 months (26).
TABLE 67-1 Infectious Complications of Transfusion of Blood or Blood Products
Transfusion-related immunosuppression with secondary infection
Postsurgical patients
Patients with burns affecting more than 10% of body surface area
Infections potentially transmitted by transfusion:
Hepatitis
HAV, HEV
HBV
HCV
HDV
HGV (GBV-C)
CMV
EBV, HHV-8
Other viral infections
HIV-1 and HIV-2
HTLV-I and HTLV-II
Other non-A, non-B, non-C agent(s) (TT virus, SEN virus)
CMV, cytomegalovirus; EBV, Epstein-Barr virus; GBV-A, B, GB viruses A and B, respectively; HAV, HBV, HCV, HDV, HEV, HGV, hepatitis A, B, C, D, E, G viruses, respectively; HIV-1, HIV-2, human immunodeficiency virus types 1 and 2, respectively; HTLV-I, HTLV-II, human T-cell leukemia virus types I and II, respectively.
The organized collection of blood and blood products for civilian use began in 1947. Hepatitis B virus (HBV) was defined serologically by 1972; its partial control has led to our understanding of other causes of PTH, including hepatitis C virus (HCV), hepatitis delta virus (HDV), hepatitis G virus (HGV), other non-A, non-B, non-C hepatitis viral agents, cytomegalovirus (CMV), Epstein-Barr virus (EBV), and, rarely, hepatitis A virus (HAV) and hepatitis E virus (HEV) (8,10,27, 28, 29, 30, 31, 32and33). The epidemic of human immunodeficiency virus type 1 (HIV-1) infections led to its recognition in the early 1980s as transmissible by blood and blood products (34). Other infectious agents transmitted by blood and blood products include protozoa, filaria, spirochetes, other viruses, and bacteria (1,8, 9and10,27, 28, 29, 30and31).
The remainder of this section reviews each of these pathogens and the prevention of their transmission by blood transfusion after a brief discussion of the recently recognized phenomenon that blood transfusion may cause immunosuppression and thereby predispose the recipient to infections. Table 67-2 summarizes the most recent donor selection criteria of the American Association of Blood Banks (AABB) for the protection of recipients of donor blood (35).
Infections after Noncontaminated Blood Transfusion
Several authors have reviewed the observations that blood transfusions may result in suppression of the recipient’s immune defenses (TRIM), leading to secondary infections (36, 37, 38and39), in addition to an association of transfusion with recurrence of malignancy and increased (renal) allograft survival (14,38,40, 41and42). Using multivariable analysis, Tartter (43) and others (44, 45, 46and47) have presented data associating blood transfusion with infection after various types of surgery. Subsequently, reports have made similar associations after surgery for trauma (48, 49, 50, 51, 52and53), Crohn’s disease (54), gastrointestinal bleeding (55), open fractures of an extremity (56), and coronary artery bypass surgery (57,58), and with healthcare-associated infections in critical care patients (59,60). Recent studies have shown that RBC transfusions have also been associated with infections, in addition to pulmonary dysfunction, TRALI, and enhanced mortality in critical care (61). Increased rates of infection after noncontaminated blood transfusion have also been documented in humans with burns affecting more than 10% of their bodies (62); initial studies in an animal burn model indicated similar findings (63,64). However, reports after elective surgery have failed to document such an association (65,66), and others have questioned the relationship of transfusion to TRIM and infection in critically ill patients (67).
The proposed mechanism accounting for these observations is immunosuppression induced by the transfusion; Waymack and Alexander (68) noted a higher incidence of tumor recurrence and reduced survival among oncologic surgery patients who received perioperative blood transfusions. Waymack et al. (69,70) suggested that the mechanism of this immunosuppression may be alteration of macrophage arachidonic acid metabolism, given elevations of prostaglandin E and a decrease of interleukin-2 (IL-2) production documented in animal models after allogeneic transfusion (69, 70and71). Lenhard et al. (72) reported decreased lymphocyte antigen responsiveness in transfused patients with chronic renal failure before renal transplantation; Lenhard et al. (73) also documented increased circulating blood monocytes with augmented prostaglandin E production in transfused patients with chronic renal failure. An analogy with the immunosuppression of pregnancy resulting from a blunted IL-2 response and upregulation of interleukin-4 (IL-4) and interleukin-10 (IL-10) has been summarized (40). These altered cytokine kinetics may lead to enhanced Th2 and depressed Th1 responses. Other authors have suggested optional mechanisms, including antigen-induced anergy and immune tolerance, and the effects of transfused cytokines (38,40, 41and42,74,75). These reported immunologic abnormalities in transfusion recipients have not been linked with potentially transfused agents (e.g., CMV). Given that the transfusion of RBC units after 14 days’ storage is more often associated with pneumonia in trauma patients (76,77), the transfusion of cytokines leading to TRIM is even more plausible because cytokine levels increase with duration of storage without LR (75).
TABLE 67-2 American Association of Blood Banks Criteria for Protection of Recipients of Donor Blood (2009)
Appearance of good health in donor, including lack of major organ disease, malignancy or bleeding tendency.
Oral temperature of donor ≤37.5°C (99.5°F); age 16 y or older, hemoglobin/hematocrit ≥12.5 gm/dL/>38%
Permanent exclusion for stigmata of injectable drug use or for use of a needle, even once, for nonprescription drug administration
Deferral of donor for 2 wk after live attenuated bacterial or viral vaccine receipt (measles, mumps, oral polio, oral typhoid, yellow fever), except for 4 wk after rubella or varicella zoster vaccine and for 1 y after rabies vaccine given for a rabies prone animal bite.
Donor deferral for 12 mo after HBIG
Donor deferral if during the previous 12 mo the donor was given blood or potentially infected blood products (components, human tissue or plasma-derived clotting factor concentrates).
Permanent donor deferral:
If history of hepatitis after age 11 y,
If HBsAg (confirmed) or anti-HBc positive (positive at two different donations),
If anti-HCV, HTLV-I/II, or HIV-1/2 seropositive,
If in a high-risk group for HIV-1/2 infection, including having sex with a person from Africa,
If prior donation led to hepatitis, HTLV-I/II, or HIV-1/2 infection in recipient
A history of babesiosis or Chagas’ disease,
Donors with a family history of Creutzfeldt-Jakob disease, donor after receipt of human pituitary growth hormone or dura-mater grafts, and donor at risk for variant Creutzfeldt-Jakob disease (e.g., receipt of bovine insulin made in United Kingdom, having spent more than 3 mo in United Kingdom between 1980 and 1996, having lived in Europe for ≥5 y, or having received blood transfusion in the United Kingdom. or France between 1980 and the present),
Having made payment for sex or having received any payment for sex, or, if the blood donor is a male, having ever had sex with another male.
Donor deferral for 12 mo after:
Unsterile body piercing or application of a tattoo or permanent make-up unless performed aseptically in state-regulated facility,
Mucous membrane exposure to blood or skin penetration by an instrument contaminated with blood or a body fluid,
Sexual exposure to a person with symptomatic viral hepatitis or confirmed positive test for HBsAg or HVC antibody,
Sexual contact with a person infected with or at high risk for HIV-1 infection,
Incarceration for >72 h in a correctional institution,
History of syphilis or gonorrhea, reactive screening test or confirmatory test for syphilis, or completion of therapy for syphilis or gonorrhea,
After return from an area endemic for leishmaniasis.
Donor deferral due to malaria (plasma donations excepted):
3-y deferral for those after recovery from malaria or for persons who lived in a malaria endemic area for ≥5 y,
12-mo deferral for travelers after return from a malaria endemic area if free of symptoms suggestive of malaria,
3-y deferral for immigrants after departure from malaria endemic areas if asymptomatic,
12-mo deferral of residents from countries that are malaria free but who have traveled to an area where malaria is endemic (acceptance as donor if symptom free).
Donor deferral until 14 d after recovery from suspected or documented West Nile virus (WNV) infection, or until 28 d from onset of illness (suspect or known to be WNV)
Donor deferral after smallpox vaccination without complications for 21 d or until scab has fallen off, whichever is longer; donor deferral after smallpox vaccination with complication until 14 d after resolution of complication
Donor is provided opportunity in confidence to declare collected blood unsuitable for transfusion by call-back system.
Anti-HBc, antibody to hepatitis B core antigen; HBIG, hepatitis B immune globulin; HBsAg, hepatitis B surface antigen; HCV, hepatitis C virus; HIV-1/2, human immunodeficiency virus types I and II, HTLV-I/II, human T-cell leukemia virus types I and II. (From Price TH, ed. Standards for blood banks and transfusion services, 26th ed. Bethesda, MD: American Association of Blood Banks, 2009.)
Current data clearly suggest that transfusion is a modulator of the immune system, although an independent role for transfusion as an immune suppressant is not yet definitive (37,40, 41and42). However, in one prospective randomized trial of postoperative infection (74), as well as other reports (39,59,60), a dose-dependent relationship of transfusion to infection risk and immune suppression has been supported.
Several methods are available for avoiding the potential immunosuppressive effects of transfusion associated with surgery and trauma. Improved surgical techniques with attention to hemostasis may avoid much of the need for transfusion. Furthermore, there is minimal scientific justification for the current hemoglobin level (10 g/dL) at which transfusion is advocated (78,79). The decreased blood viscosity associated with reduced hemoglobin concentrations of anemia may increase cardiac output and partially compensate for decreased oxygen delivery to tissues (79,80). Acceptance of reduced indications for transfusion according to hemoglobin concentration and the use of autologous blood for transfusion (self-donated preoperatively or intraoperatively recovered blood) will also reduce the immunosuppressive effects of transfusion (79). Furthermore, restrictive transfusion strategies in critical care and patients with acute coronary syndromes have reduced TRALI and mortality (81, 82, 83and84). Hebert et al. have reported noteworthy benefits from a restrictive transfusion strategy (hemoglobin was maintained from 7-9 g/dL) in critical care patients at 25 Canadian hospitals; observed reductions were seen in ICU and hospital mortality, organ dysfunction, and length of ICU and hospital stay (85). Restrictive transfusion criteria are now being advocated for various types of surgery, after trauma, and in critical care and are advocated by the Society of Critical Care Medicine in its latest guidelines (82,86,87,88,89,90,91). In the past, prior to near-universal prestorage LR, other preventive techniques have included the use of leukocyte-reduced blood products, such as frozen deglycerolized RBCs or RBCs after treatment with second-generation micropore or thirdgeneration absorption blood filters, and the use of blood alternatives (e.g., hemoglobin solutions depleted of erythrocyte stroma, chemically modified hemoglobin solutions, and artificial RBCs or neohemocytes) (14,92, 93, 94, 95, 96and97). These leukocyte-reduction techniques also prevent many febrile transfusion reactions (97). The stimulation of the patient’s bone marrow with erythropoietin to produce RBCs is another method to avoid transfusion and has been effective in dialysis patients, despite side effects (98).
The concepts of immunomodulation and increased infection risk after allogeneic blood transfusion have been unified mechanistically through the appreciation of cytokine release by leukocytes during blood storage (16,41,42,44,75). A number of studies have demonstrated that leukocyte-depletion of transfused RBCs may have favorable effects on infection rates, morbidity, and/or mortality (75,99, 100, 101and102). These studies further support the role of cytokines, in a variety of clinical settings, derived from leukocytes as a cause of described ill effects of transfusion. Additionally, while LR of RBCs is not universally accepted (103,104), LR programs have reduced infection risks post operationally and after trauma (75,105, 106, 107, 108and109). Much of the confusion about the benefit of LR of all donor units stemmed from the lack of understanding of the need for prestorage LR to avoid cytokine accumulation during storage (110). The ARC has implemented LR prestorage for all donor units since 2000.
Infections after Transfusions Contaminated with Pathogens
Febrile reactions, defined as a temperature rise of 1°C (2°F) or more, may be associated with 1% to 2% of all RBC transfusions (3,13,97). In addition to an infective cause, either ongoing in the recipient or rarely resulting from bacterial contamination of the transfused blood product (see later discussion), fever may also follow a hemolytic transfusion reaction or may be associated with cytokines or antibodies in the transfused blood products, or antibodies in the recipient against leukocyte or platelet antigens (16,97). Such febrile, nonhemolytic transfusion reactions may present as acute noncardiogenic pulmonary edema resulting from either reactive lipid products or antileukocyte antibodies or in association with the platelet refractory state (failure of the platelet count to rise after transfusion because of rapid antibody-mediated clearance). These febrile reactions are most commonly seen in multiply transfused, alloimmunized recipients, in multiparous female recipients of transfused blood or blood products, or after transfusion of blood or blood products from multiparous female donors. These reactions can be avoided by using leukocyte-reduced blood products (3,9,16,21,42,97). Febrile antiplatelet reactions may resolve with LR, but the platelet refractory state is seldom benefited.
Another febrile reaction related to transfusion of immunoincompetent and, rarely, normal hosts is a delayed reaction occurring 1 to 2 weeks after transfusion with the presentation of fever and erythroderma (111,112). This often fatal transfusion-associated GVHD is not infectious, is usually not confused with a febrile transfusion reaction, and is obviated by irradiation of cellular blood products for at-risk patients (3) and by universal LR (21).
The most frequent serious transfusion complication is the transmission of infection, of which hepatitis and, more recently, HIV-1 are the most important. Parenteral transmission of hepatitis was not recognized until 1883 when an outbreak occurred among recipients of a smallpox vaccine of human origin (113). In 1938 and again in 1942, a yellow fever (YF) vaccine stabilized with human serum was reported to have caused jaundice among recipients (114,115); a virus was presumed to have contaminated the human serum. Subsequently, epidemiologic studies and human volunteer experiments defined two forms of viral hepatitis: hepatitis A or infectious hepatitis, believed to be transmitted only orally (short incubation period of 15-50 days), and hepatitis B or serum hepatitis, associated with parenteral exposure (long incubation period of 50-180 days; 116). Although it was known that both forms of hepatitis could be transmitted parenterally by blood and that hepatitis A could be acquired orally from various body fluids, physicians identified the form of hepatitis by exposure history until 1965 when Blumberg et al. (117,118) serendipitously associated Australia antigen with the surface antigen of hepatitis B. The development of serologic assays for hepatitis B and subsequently for hepatitis A opened the door to our evolving understanding of the other agents of PTH (119, 120and121; Table 67-3).
TABLE 67-3 Established Posttransfusion Hepatitis Viruses
CMV, cytomegalovirus; EBV, Epstein-Barr virus, HAV, HBV, HCV, HDV, HEV, HGV, hepatitis A, B, C, D, E, G viruses, respectively; ND, not defined; NANB, non-A non-B; NANBNC, non-A non-B non-C.
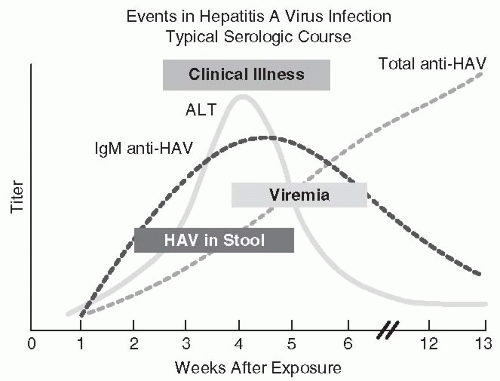
Hepatitis A Much of the epidemiologic information contrasting hepatitis A and hepatitis B resulted from human volunteer studies performed in the 1940s (116) and at the Willowbrook State School in New York between 1956 and the late 1960s (122,123). These and other studies defined the differences of incubation periods and antigenicity, the presence of HAV in feces, and the lack of chronic carriage of HAV (116,122, 123and124). HAV was first visualized in stool using immune electron microscopy in 1973 (125); this finding ultimately led to methods for detection of serum antibody to HAV. The periods of viremia, clinical illness, and aspartate aminotransferase elevation have been related (Fig. 67-1); the transience of the immunoglobulin M (IgM) antibody response, followed by a persisting immunoglobulin G (IgG) response, has also been defined by both radioimmunoassays (RIAs) and enzyme immunoassays (EIAs) (126, 127, 128and129). Although IgM antibody usually disappears within 3 to 6 months after infection, it may persist for more than 300 days in 10% to 15% of patients (130).
FIGURE 67-1 The clinical, virologic, and serologic course of acute hepatitis A virus infection. (Redrawn from http://www.cdc.gov/hepatitis/Resources/Professionals/Training/Serology/gr_hav.htm)
Because of the lack of an HAV carrier state and the brevity of HAV viremia (usually 2 weeks or less, with onset of viremia 7-10 days before onset of clinical symptoms, Fig. 69-1), frequent episodes of PTH A are unlikely (9,26,120,121,131,132). With few exceptions, this has been the case (133, 134, 135, 136, 137, 138, 139, 140and141). Usually, PTH A occurs as a sporadic case report after blood donation during the incubation period of the illness (139, 140and141). Unfortunately, several outbreaks have been due to single contaminated units of blood being transfused to multiple infants, resulting in nursery outbreaks with secondary and tertiary cases (134,136,137,139). One outbreak occurred among cancer patients treated with IL-2 and lymphokine-activated killer lymphocytes apparently resulting from contaminated serum in the lymphocyte culture medium (138); another outbreak has occurred among patients with hemophilia given clotting factor concentrates inadequately treated to inactivate HAV (142). These few reported outbreaks might have been prevented by serologic testing for antibody against HAV; however, the 50% seroprevalence rate for HAV IgG antibody among Americans by age 50, the possible persistence of IgM antibodies for 3 to 6 months after infection despite lack of infectivity, the frequency of symptoms during the viremic phase of the illness (123), and the rarity of fatal illness resulting from HAV are strong arguments against the economic or the medical merit of routine testing of donor blood for HAV antibody (9,27,132,143). The estimated current residual risk of acquiring HAV from a unit of transfused blood is estimated at 0.0001% (9,144). The prompt administration of immune serum globulin (ISG) prophylaxis would be appropriate for a recipient of blood found after transfusion to contain HAV (27). The elimination of febrile symptomatic patients as blood donors generally prevents HAV transmission. Frequent recipients of clotting factor concentrates are candidates for receipt of HAV vaccine (30,142).
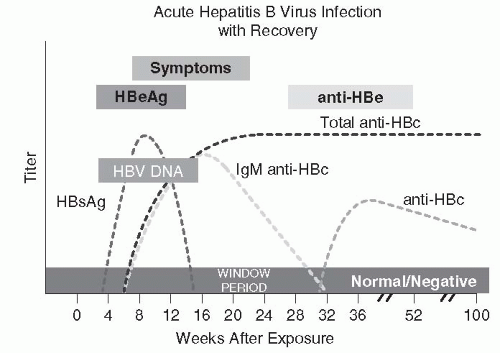
Hepatitis B Blumberg et al.’s classic seroepidemiologic studies of diverse populations led to the discovery of a unique antigen in Australian aborigines and recipients of multiple transfusions, which was called “Australia antigen” (117,118,145,146). The terminology subsequently evolved from hepatitis-associated antigen to hepatitis B antigen and finally to hepatitis B surface antigen (HBsAg) when it was associated with the surface lipoprotein of HBV (147,148). HBsAg can be found in serum of patients developing acute hepatitis B for 30 to 60 days before illness and may persist for variable periods after clinical recovery (Fig. 67-2). Persistence for longer than 6 months is defined as the chronic carrier state. Antibody against HBsAg (anti-HBs) develops as HBsAg disappears and accounts for long-term immunity (149).
FIGURE 67-2 The clinical and serologic course of hepatitis B infection. (Redrawn from http://www.cdc.gov/hepatitis/Resources/Professionals/Training/Serology/gr_hbv_acute.htm)
Hepatitis B core antigen, reflecting active viral replication, transiently appears in the blood during acute infection, only to be replaced by its antibody (anti-HBc) (150). Anti-HBc appears during acute infection as an IgM antibody and is replaced in up to 80% of patients by a persistent IgG antibody during convalescence (151) (Fig. 67-2).
Hepatitis B e antigen (HBeAg) is a soluble product of HBV infection found transiently in serum during acute hepatitis B and in the serum of patients with chronic hepatitis (152, 153and154) (Fig. 67-2). The presence of HBeAg correlates with the presence of HBV virions in serum. Antibody to HBeAg is more commonly found in chronic asymptomatic carriers of HBsAg (153,154). The presence of HBeAg is associated with increased risk of maternal-fetal HBV transmission and transmission via accidental needle stick (155, 156and157).
Transmission of HBV occurs through percutaneous or transmucosal inoculation of HBV in blood or infectious body fluids (primarily semen and breast milk). Inoculation may occur by contaminated needles, during sexual contact, at birth, or during transfusion. Continuous household or institutional contact with an infected person may presumably transmit infection via unapparent exposures.
Acute hepatitis B causes a chronic viral carrier state in 6% to 10% of infected adults and 90% of infected newborns with or without chronic hepatitis (25% of carriers). The most serious sequelae in chronic carriers of HBsAg are cirrhosis and hepatocellular carcinoma. It is estimated that approximately 800,000 to 1.4 million chronic HBsAg carriers live in the United States as of 2007 (158). These individuals serve as a potential reservoir within the pool of blood donors. High-risk groups for chronic carriage of HBV are shown in Table 67-4.
TABLE 67-4 High-Risk Groups for Acquiring Hepatitis B Infection
Before the development of assays for HBsAg, it was believed that HBV accounted for most cases of PTH. When use of the first-generation immunodiffusion assays for HBsAg was initiated voluntarily on donor blood in 1969 and became mandatory in 1972 (Table 67-5), it was anticipated that PTH would be virtually eliminated. Although there were marked reductions in the frequency of PTH (30-55%) and mortality related to transfusion, the problem persisted. Hepatitis B may still account for up to 10% of cases of PTH (9,159,160). Recently published risk estimates for PTH resulting from HBV are 1 infected unit per 220,000 donor units (0.00045%; 144,161); among first-time blood donations, 19.14 units are positive for HBsAg per million units donated (162).
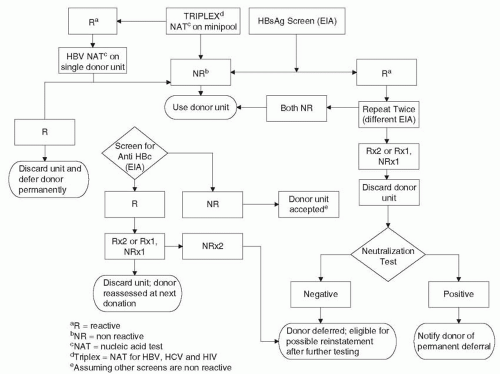
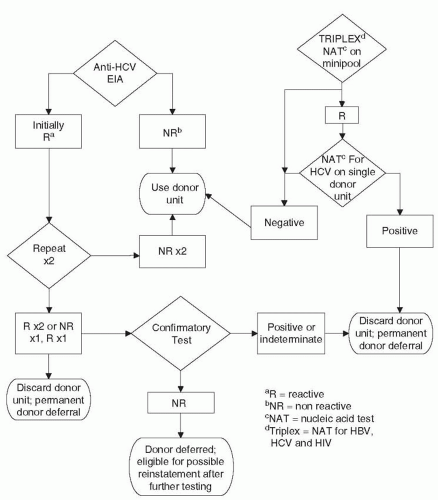
The transmission of HBV via transfusion has been reduced to present levels because of the screening of all blood donors for HBsAg with more sensitive assays. Counterimmunoelectrophoresis was introduced in 1972 to 1973, and sensitivity for HBsAg detection was further increased by currently available RIAs, reversed passive hemagglutination, chemiluminescence immunoassays, and EIAs (159,160,163, 164, 165and166). With the successive introduction of second-generation counterimmunoelectrophoresis and current third-generation tests for HBsAg (Table 67-5), Alter et al. (167, 168and169) documented the parallel reduction of PTH resulting from HBV. This reduction was likely augmented because of current ARC donor selection and deferral procedures and the elimination of paid donors in favor of all-volunteer donors (9,10,159,160,167, 168, 169and170). HBeAg has been found more frequently in paid blood donors (15%) compared with volunteer donors (5%; 171). Further reduction of PTH due to HBV may also result from the fact that asymptomatic HBsAg carriers continue to be removed from the donor pool through repeat donor testing (27,172).Figure 67-3 presents the current algorithm for screening donor blood for HBV.
TABLE 67-5 Serologic Tests for Infectious Agents Performed on Blood and Blood Products before Transfusion
gNAT, nucleic acid testing (polymerase chain reaction—PCR, or transcription-mediated amplification—TMA); 6-16 minipool or single donor testing currently utilized.
hBacterial detection—culture or alternative detection method (CO2 production, O2 consumption, fluorescent labeling, pH > 6.4). ALT, alanine amino transferase; anti-HBc, antibody to hepatitis B core antigen; anti-HCV, antibody to hepatitis C virus; ChLIA, chemiluminescence immunoassay; CIE, counter immunoelectrophoresis; EIA, enzyme immunoassay; HBsAg, hepatitis B surface antigen; HIV-1/2, human immunodeficiency virus types 1 and 2; HTLV-I/II, human T-cell leukemia virus types I and II; RIA, radioimmunoassay.
The residual frequency of PTH resulting from HBV is apparently due to the fact that HBsAg is circulating at undetectable levels for current screening assays; some of these donor units can be eliminated by screening for anti-HBc (173, 174, 175, 176, 177and178). Such donor unit screening was initiated in 1986 (Table 67-5) as a surrogate test for non-A, non-B PTH but is believed to have contributed further to the reduction of HBV-related PTH. It is estimated that because of the institution of surrogate screens for non-A, non-B PTH (anti-HBc and alanine aminotransferase [ALT], the latter no longer performed), the incidence of HBV-associated PTH has further decreased by up to 84% to current levels (144,179). However, this reduction may also have been affected somewhat at that time (and currently; 35) by more stringent donor population screening to prevent transfusion-related HIV-1 transmission (159), because the incidence of PTH was already further dropping before the initiation of anti-HBc screening (180). Regardless, the risk of HBV-related PTH has currently dropped at least to 0.00045% per transfusion recipient (8,146,159). However, further reduction may require more sensitive assays, because 4% to 12% of HBV-DNA carriers are identified by nucleic acid testing (NAT) and are seronegative for HBsAg, HBcAb, and other HBV serologic markers (30,181). Such NAT positive but HBV serologically negative donor units may not transmit HBV infection (181). HBV mutations or genotypes may contribute to falsely negative HBsAg serologic tests (8,30,182).
In addition to the marked reduction of PTH caused by HBV resulting from HBsAg and anti-HBc testing of each donor unit of blood, the development of the NAT for whole blood (to assay for HBV DNA in plasma of donors without HBsAg) has the potential to decrease the already low rate of PTH resulting from HBV (183,184). It is projected that an additional 81 HBV infected units of blood would be detected annually among the 12 million screened units (8), thereby potentially reducing the risk of HBV transmission due to transfusion by 42% to 0.00045% per unit (8,9,144,172). NAT screening for HBV was introduced for all US blood donor units between 2006 and 2010 as a triplex screen with HCV and HIV-1 (Table 67-5). The clinical benefit of NAT for HBV remains controversial (185). The addition of NAT for HBV, while expected to avoid 9 to 37 HBV transfusion-transmitted infections annually, adds an additional $39 to $130 million dollars per year to donor unit screening costs (186).
FIGURE 67-3 Algorithm for screening of donor blood for hepatitis B.
If it were established within 1 to 2 weeks of receipt that a patient had been administered a unit of HBV-contaminated blood, there are no data that the use of HBV vaccine or hepatitis B immune globulin would be of value. In this unusual situation, one could argue for the use of both preparations, as after accidental parenteral exposure, in an attempt to modify the anticipated illness. When it can be anticipated that a person is going to receive multiple future transfusions (e.g., a hemophiliac), HBV vaccine should be administered as early in life as possible. However, the mainstays of prevention of PTH B for most patients are deferral of high-risk donors and serologic testing and NAT of donor units. Given the recommendations of the Immunization Practices Advisory Committee for universal childhood HBV vaccination, the risk of PTH resulting from HBV should become even smaller (187). Before NAT for HBV, the risk of transmission of HBV via transfusion had remained stable in the United States (9,10,172). This stability and the lack of an increasing relative risk of HBV transmission probably reflect an effect of universal childhood immunization, in addition to continued refinement of donor deferral criteria and use of antigen and antibody testing and possibly NAT.
Delta Hepatitis Delta agent hepatitis was initially described in 1977 by Rizzetto et al. (188) in Turin, Italy, and was reviewed by Hoofnagle in 1989 (189). Although endemic to southern Italy, this virus has a worldwide but geographically variable distribution, including the Middle East and parts of Africa and South America. In nonendemic areas, such as the United States and Western Europe, the delta virus is found primarily in injectable drug users and multiply transfused patients, including hemophiliacs (190). HDV is a defective RNA virus that replicates only in the presence of HBV with circulating HBsAg (189). It is composed of an inner low-molecular-weight RNA genome associated with the internal delta antigen protein and coated with HBsAg as the surface protein (191).
HDV infection occurs in only two settings: as a simultaneous coinfection with acute HBsAg-positive hepatitis B or as a superinfection superimposed on the chronic HBsAg carrier state (189, 190and191). During coinfection, although the ensuing hepatitis may be severe, biphasic, and protracted with a 2% to 20% mortality rate, most patients recover as hepatitis B resolves and fewer than 5% of patients develop chronic hepatitis (190,192). The mortality rate resulting from coinfection of HDV with HBV contrasts with the <1% mortality rate associated with hepatitis B alone (189). Illness associated with HDV is defined by a resurgence of the ALT serum levels after an initial decline, concomitant with the appearance of a transient anti-HDV-IgM response and followed by the development of low titer anti-HDV-IgG (189,192). These antibody responses may be detected by commercially available RIAs and EIAs (193,194).
In contrast to acute coinfection, when HDV infection is superimposed on the chronic HBsAg carrier state, most patients (70%) develop chronic hepatitis with continued presence of HBsAg and HDV in the serum (192). Sixty percent to seventy percent of patients with chronic delta hepatitis develop cirrhosis, and most of these die from liver disease (195). Chronic HDV infection is documented by the persistence of anti-HDV-IgM in high titer (192). HDV antigen may also be detected in the liver.
Because HDV is usually parenterally spread, its frequency in PTH associated with HBV (HBsAg positive) has been evaluated; 3.5% of 262 patients with PTH resulting from HBV were positive for anti-HDV (196). Of these patients, 2.5% of those with self-limited disease were anti-HDV positive, whereas 14.5% of those with fatal hepatitis were infected with HDV. These data raise serious concerns for transfusion recipients; however, screening for HBsAg and NAT in each donor unit provides a high degree of protection for HBsAg-negative blood donor recipients (196). HDV antibody screening is not needed. However, the HBsAg-positive prospective transfusion recipient is at some risk, especially if multiply transfused. In addition to the usual HBsAg screen of donor blood, blood-derivative recipients who are HBsAg carriers should be given products from single or minipool plasma sources (196). Furthermore, all donors whose ALT is known to be elevated should be eliminated as blood sources for HBsAg-positive recipients.
Hepatitis C and Non-A, Non-B Hepatitis After the introduction of testing of all donor blood for HBsAg, it quickly became apparent that not all PTH was due to HBV (27,120,121). Hepatitis A was also quickly excluded as a potential cause of the residual PTH (120,131), as were CMV and EBV (120,197,198). It was concluded that another virus (or viruses) accounted for most of PTH cases in the United States, initially designated “non-A, non-B or type C PTH” (199,200). Much of the epidemiologic description of non-A, non-B hepatitis is now applicable to HCV, which caused most cases of non-A, non-B PTH (201, 202, 203and204).
HCV is believed to be the most common cause of nonalcoholic liver disease in the United States. The prevalence of HCV in the US population is approximately 1.8%; 73% of patients with chronic infection have genotype 1, with the remaining predominately genotypes 2 and 3 (205). The risk factors for infection somewhat parallel those of HBV. Among well-defined factors, transfusion (5-10%) and injectable drug use (60%) have accounted for most infections; transfusion is a virtually eliminated risk today (1,8, 9, 10, 11and12,172,202,205, 206and207). The risk of transfusion-related HCV infection declined between 1981 and 1988 from 17% to 6% before antibody screening (208). Antibody to HCV is found in up to 85% of injectable drug users (209,210). Other high-risk groups include prisoners, patients with transfusion-dependent bleeding disorders, and hemodialysis patients (207,209, 210, 211and212). Sexual, household, and perinatal transmission and receipt of intravenous immune globulin are less important risks for HCV (207,209,213, 214and215); sporadic cases without defined exposure have declined (from 40% to 50%) to 5% of new cases (207,208,216). In the contemporary era of home intravenous infusion therapy, risks for HCV infection previously recognized in other sites have emerged in the household setting; a recent report suggests that transmission of HCV from an HCV-infected mother to her hemophiliac child may occur via accidental percutaneous (ungloved) needle stick during venipuncture for clotting factor infusion (217). A family history of liver disease and prior history of blood transfusion, tattooing, sexual promiscuity, injectable drug use, intranasal cocaine use, and male ear piercing have been associated with anti-HCV positivity among blood donors (218,219).
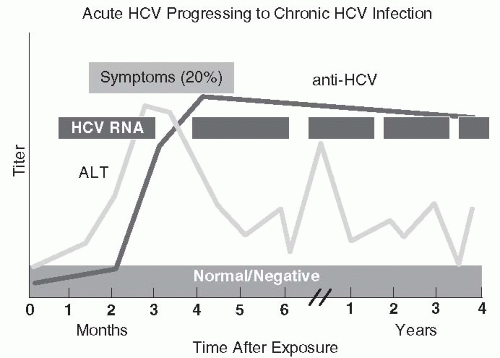
FIGURE 67-4 The clinical, biochemical, and serologic course of hepatitis C infection. (Redrawn from http://www.cdc.gov/hepatitis/Resources/Professionals/Training/Serology/gr_hcv.htm)
Approximately 75% of cases of HCV are subclinical, but, when symptomatic, hepatitis C is clinically and biochemically identical to other forms of hepatitis (201,207).Figure 67-4 depicts the clinical, serologic, and biochemical course of HCV infection. Up to 85% of patients with hepatitis C develop chronic hepatitis (31,201,220,221). Despite resolution of hepatitis after HCV infection, HCV RNA can often be detected by PCR, indicating HCV persistence in asymptomatic, biochemically normal patients (about 30% of chronic carriers; 207,220). Symptoms or serum ALT level do not correlate with disease severity (220,222). Most patients develop chronic active hepatitis with or without cirrhosis (220, 221, 222, 223and224). Cirrhosis may variably appear in 20% to 50% or more of patients (201,207,224). Chronic hepatitis C has also been linked with hepatocellular carcinoma (207,225, 226and227). Patients seropositive for HCV antibody but with normal ALT values, no HCV RNA in serum, and normal hepatic histology, although uncommon, (up to 15% of those infected) probably have recovered from HCV infection (221).
There are several studies assessing the outcome of PTH resulting from HCV. Seeff et al. (228) reported a study of long-term mortality after non-A, non-B PTH in 568 patients matched with two control groups, both of which comprised patients who had been transfused but had normal ALT values after transfusion. After an average of 18 years of followup, there was a small statistically significant increase in deaths resulting from liver disease in the patients with PTH (3.3% vs. 2.0% and 1.1% in the control groups). In a retrospective study, Tong et al. (229) defined that PTH resulting from HCV evolves into a progressive disease; chronic active hepatitis (23%), cirrhosis (51%), and hepatocellular carcinoma (5.3%) are noteworthy sequelae. Goedert et al. (230) recently reported 137 hemophiliac patients with end-stage renal disease (ESRD) and HCV infection; ESRD was significantly associated with HIV-1, older age, HBV coinfection, and a low CD4 cell count. Another study has suggested a more benign outcome (45% viral clearance by PCR) in children infected via transfusion (231).
Before the development of the current serologic tests to detect HCV, surrogate markers for non-A, non-B hepatitis were used as screening methods for donor blood. Initially, ALT assays were proposed to help reduce non-A, non-B PTH (232, 233and234). Up to 45% of donor units implicated in transmission of PTH have ALT elevations >60 IU/L (179). Discarding units of blood positive for anti-HBc was subsequently shown to reduce posttransfusion non-A, non-B hepatitis (177,178; Fig. 67-5). Approximately 53% of blood donors implicated in PTH and positive for anti-HCV have anti-HBc (179). The use of these assays instituted in 1986 and 1987, respectively, led to an approximately 30% to 40% reduction of PTH (179). After 1985, blood transfusion as a source for acquiring HCV decreased to only 4% of new cases (220).
Contemporary molecular biologic techniques led to the discovery of HCV. Nucleic acid extraction of infectious chimpanzee plasma led to the isolation of viral RNA, its transcription to DNA, and expression after insertion into a phage. Screening of several million DNA sequences for production of proteins that reacted with antibody in the serum of patients with non-A, non-B PTH led to the discovery of a polypeptide (C100-3) that was developed as an antibodycapture RIA for HCV (204). This assay was quickly shown to detect 65% of donor blood capable of transmitting chronic PTH and 17% of acute PTH (235). This commercially available antibody capture RIA was adapted also to an EIA, which has been shown to detect HCV as a cause of PTH (236, 237and238). Initial enthusiasm for this assay dampened because of the delayed seroconversion and, therefore, potential seronegativity of blood donors infected with HCV (despite the greater sensitivity of first-generation EIAs compared with RIA; 210, 239, 240, 241and242). The sensitivity of the EIA and RIA for preventing non-A, non-B PTH resulting from HCV was variably estimated at 60% to 85% (206,239,243). First-generation assays for anti-HCV became positive up to a year after acute hepatitis C, and up to 20% of patients remained seronegative by these assays due to the insensitivity of the first-generation screening assays (210,237,244). Regardless, contrasting the period when only surrogate tests for non-A, non-B PTH were used to the period after 1990 when the first-generation assays were implemented, Donahue et al. (245) demonstrated an 84% reduction of the risk of PTH resulting from HCV among transfused cardiac surgery patients (Table 67-6).
FIGURE 67-5 Algorithm for screening of donor blood for hepatitis C and hepatitis non-A, non-B, and non-C.
TABLE 67-6 Estimated Transmission Risk of Various Potentially Transfusion-Transmitted Infections, USA (Based on 2008 Data)
The ability to prevent PTH resulting from HCV was further improved by second-generation EIAs, which incorporated detection of antibodies to the core antigen (C22-3 protein) and the C200 antigen, which combines the epitopes included in the c33c and C100-3 proteins (coded by the NS-3 and the NS-4 regions of the RNA genome) (30,206). Wang et al. (246) demonstrated 100% sensitivity of a second-generation EIA incorporating these three antigens, compared with 83% sensitivity of the first-generation assay; the newer assay also detected anti-HCV 6 weeks earlier than the single-antigen EIA. Using dot-blot assays, several groups demonstrated that antibodies to the c33c or core proteins (C22) consistently appeared before those to C100-3, from 4 to 13 weeks after transfusion (247,248).
In addition to their sensitivity, another concern with the first-generation anti-HCV EIAs and RIAs was their positive predictive value. A positive EIA or RIA was confirmed by the more specific recombinant immunoblot assay (RIBA) in only 19% to 60% of cases (243,249, 250, 251and252), with only one study showing 100% correlation of the screening EIA with RIBA (239). Newer RIBA and matrix (a semiautomated immunoblot) assays each contain four recombinant antigens (253). In keeping with Bayes’ theorem, in highrisk populations, 70% to 100% of sera repeatedly reactive by second-generation EIAs were determined to be true positives by the RIBA or matrix assays, whereas fewer than 50% of repeatedly reactive second-generation EIAs are true positive in low-risk populations (220,253).
Despite the limitations of first-generation EIAs, on May 2, 1990, an anti-HCV first-generation screening assay became mandatory on all donor blood (Table 67-5). Blood banks began using the second-generation EIA to screen for HCV antibody on April 6, 1992. Because of the previously discussed suboptimal sensitivity of these assays for HCV infection in a donor unit, the screening of donor blood for ALT level and anti-HBc was considered as important and was retained as required assays until June 20, 1995, when the requirement for ALT assays was rescinded (Table 67-5 and Fig. 67-5) (29,35,206,243,249,250,254).
Third-generation EIAs were initially tested in Europe and then approved by the FDA in May 1996 for use by blood donation centers. Similar to the second-generation assays, these third-generation assays incorporate recombinant antigens and include those described previously for the second-generation products, in addition to the protein product of the NS-5 region of the genome (RNA polymerase) (30,255,256). These assays detect all genotypes and offer marginal improvement in reducing the seroconversion window (by 12 days) and the potential infectivity of a donor unit (15% reduction of an already low rate) (30,255,256). All HCV-antibody positive specimens by EIA are confirmed as positive by RIBA; a second-generation RIBA now incorporates the same recombinant antigens as the newer EIAs.
The risk of PTH was estimated to be <0.5% per patient transfused with the use of the third-generation EIA (30,220,253). In addition to screening for HBsAg and antibody to HIV-1 and the adoption of a totally volunteer blood donor system with donor screening for HIV-1 infection risks, screening for ALT, anti-HBc, and especially anti-HCV had a major impact on the reduction of PTH. Donor questionnaire screening and donor unit serologic screening produced an overall low prevalence of anti-HCV positivity among volunteer blood donors (<0.5%) (210,239,240,243,249,253). The frequency of anti-HCV positivity has been found to be higher among paid blood donors (10%; 257), compared with volunteer groups (0.36%); anti-HCV positivity has been estimated to be 53.6 donor units positive for each million units collected (162). Despite this low prevalence in volunteer donors, anti-HCV screening has demonstrated an impact (Table 67-6) because of the frequency of PTH (58-95%) after transfusion of anti-HCV reactive donor units and because of the ability of second- and third-generation EIAs for anti-HCV to detect 90% or more of donor units that transmit HCV (206,210,237,239,241,243,253). The residual risk with the third-generation EIA was related to the possible 12- to 14-week window period after infection with HCV before the appearance of antibody (258).
With these added screening tools, but before NAT, the risk of PTH had declined to <0.5% of transfused patients, a risk comparable with or less than the risk of hepatitis in nontransfused hospitalized patients (202,206,253). Furthermore, 58% to 80% of the patients developing PTH in the past have developed persistent ALT elevations and 26% to 85% have chronic hepatitis; 90% of these patients are anti-HCV reactive (179,188,198,215,219,231). Therefore, with current blood donor unit serologic screening, there is the potential not only to reduce PTH resulting from HCV by 90% or more but also to reduce chronic hepatitis resulting from HCV after transfusion by 80% to 90% (235,253). On June 20, 1995, the prior requirement for ALT assays on all donor units was made optional because of rejection of many acceptable donors, especially males, because ALT assays are subject to interlaboratory variation and because the risk of PTH in groups receiving blood screened with or without ALT testing was equivalent (254).
The use of NAT has further enhanced the ability to decrease the already low frequency of PTH resulting from defined HCV infection in donor blood (207,259, 260, 261and262). PCR can detect HCV RNA within 59 days of infection (8). With resolution of PTH resulting from HCV, HCV RNA detected by PCR has been shown to clear from blood as antibody levels decrease (248). Before NAT for HCV, the risk of PTH resulting from HCV had been reduced to approximately 0.001% per unit of blood (8,9). The addition of NAT in 1999 (initially as a duplex screen with HIV-1, and now a triplex screen also with HBV) has further reduced this risk to 0.00005%, an estimated 72% further risk reduction (8,9,144,172,263). Despite NAT, HCV can still be transmitted by anti-HCV- and NAT-negative blood products (264). However, Dodd et al. (172) have documented the very low risk of HCV hepatitis after transfusion; they reported only 74 units of blood from more than 19 million drawn between March 1999 and February 2002 that were HCV antibody negative but HCV-RNA positive, for a rate of one positive unit per 267,700 units. This low rate has been confirmed by two more recent studies, one of which assessed 66 million donor units between 1999 and 2008 (185,265). In addition, the Centers for Disease Control and Prevention (CDC) has reported no transfusion-related HCV seroconversions among 11,171 hemophiliac patients between May 1998 and June 2002 and no transfusion-related cases during 2007 (132,266). These improved safety data reflect an estimated decrement of the window period of infectivity of donor blood for HCV from 82 days with current antibody tests alone to 23 to 36 days by also incorporating NAT (8,267). NAT has been estimated to avert 56 to 59 HCV infections annually at a cost of several hundred million dollars annually (186). PTH has become an uncommon event, reflecting the current safety of the blood supply.
Additional potential methods to prevent PTH resulting from HCV await development of a vaccine. Studies evaluating pretransfusion or post-transfusion administration of ISG have demonstrated conflicting results (268, 269and270). The variable efficacy probably reflects inconsistent anti-HCV content of preparations. ISG is not given in this setting (207).
Other unsolved issues regarding non-A, non-B PTH remain. Only 91% of non-A, non-B PTH is associated with anti-HCV reactivity (206,209,237). Although the incubation periods for anti-HCV-positive and anti-HCV-negative PTH are the same (6-12 weeks) (244), anti-HCV-positive patients are more seriously ill and have twice the incidence of chronic hepatitis (206,244). Other agents remain yet to be defined to account for the 9% of cases of PTH that are not due to HAV, HBV, or HCV (8, 9and10,172,244,271,272).
Hepatitis E Hepatitis E virus (HEV) is a single-stranded RNA virus in the Hepeviridae family. The epidemiology of HEV is similar to HAV in terms of fecal-oral transmission; HEV is more common in developing countries in tropical areas (144). The seroprevalence is <2% in the United States. Pigs, chickens, and other animals may be infected with HEV and serve as a source of human infection (273). Illness is more severe than HAV infection, with 2% fatality (higher in pregnancy; 144). There are rare reports of transfusionrelated infection, both in developing and developed countries (33,274,275). Although HEV is a recently recognized pathogen, with our understanding of HEV yet evolving, as with HAV, there is not a need at this time to consider screening for HEV given its lack of chronic infection and likelihood of donor self-deferral during acute illness.
Hepatitis G Similar to the original work with HCV, in 1996 Linnen et al. (276) reported the cloning of HGV from the plasma of two patients, one with non-A, non-B hepatitis and the other asymptomatic with intermittent enzyme elevations. This group reported that HGV was genomically similar to another human virus isolate from a surgeon ill with hepatitis in the 1960s, termed “GB virus C.” HGV is a member of the Flavivirus family, along with HCV and GB virus C. Using reverse transcriptase PCR technology, 2 of 12 patients with PTH were found positive for HGV RNA by Linnen et al. (276), as were 5 of 38 patients with non-A to non-E community-acquired hepatitis. Four of the latter five patients remained HGV-RNA positive for 2 to 9 years without evidence of chronic hepatitis. In addition, Linnen et al. reported 13 of 779 (1.7%) of volunteer blood donors with normal ALT values positive for HGV RNA, as were 11 of 709 donors (1.5%) with ALT elevations. These authors reported HGV to be globally distributed. More recent screening studies have confirmed these findings (277).
Subsequent studies have confirmed this work and have found HGV in hemodialysis and postoperative patients, presumably transfusion associated (278, 279and280). Several authors have reported on the clinical disease associated with HGV, whether alone or in association with HCV; HGV is at worst a cause of mild acute hepatitis (280,281), but frequently infection with HGV is asymptomatic without evidence of hepatitis (280, 281and282). HGV does not augment disease when accompanying HCV (280, 281, 282, 283and284). HGV was not associated with chronic hepatitis (279,280,282). HGV is persistent in HIV-1 infected men and had variable effects on short- and long-term survival (285). HGV is also prevalent in injectable drug users and hemophiliacs; HGV can be passed vertically mother to child and after heterosexual or homosexual contact with HGV-positive partners (280). Its modes of transmission parallel HCV generally. Tanaka et al. (283) reported HGV responsiveness to interferon-a, but most patients relapsed.
Despite the prevalence of HGV in the volunteer blood donor population (1-4%), its role in PTH remains to be determined, because 75% of patients with transfusionacquired HGV lack biochemical evidence of hepatitis (280,281). Those with hepatitis have only mild elevations asynchronous with their HGV-RNA levels (281). The relevance of HGV (and GB virus C) to PTH remains to be defined with broader seroepidemiologic, biochemical, and clinical studies. Currently, there appears to be no reason to test blood donors for HGV (286,287).
Cytomegalovirus CMV is a member of the herpes virus family of DNA viruses (human herpes virus 5, HHV-5). Like other members of this group of viruses, latency is the rule after recovery from acute infection. Acquired CMV infection rarely presents as hepatitis, but may do so in adult patients (288). Epidemiologically, CMV is acquired by human-to-human contact, congenitally or perinatally from mother to child by contact with cervical secretions, postnatally by an infant via breast milk, and in settings for the care of multiple children (e.g., in neonatal nurseries, in day care centers, and in the family setting) (289). CMV is also transmitted by heterosexual or homosexual contact, by transfusion of donor blood, and by transplantation of donor organs (289).
CMV infections are classified as primary (with seroconversion from negativity to positivity), reactivation of an endogenous infection, or reinfection with a new exogenous strain of CMV (in a seropositive host) (288,289). The latter two forms of infection can be distinguished by using restriction enzyme DNA analysis, but this is not routinely practical (290). Because there are no accurate data on the proportion of reactivation and reinfection for CMV posttransfusion infections, these two forms of infection are called “recurrent infections” (288,289).
Depending on socioeconomic stratum and age, the seroprevalence of CMV ranges from 25% to 88% (144, 287, 288, 289, 290, 291, 292, 293and294). In studies of CMV infection after transfusion in normal hosts before 1972, the incidence varied from 16% to 67% (288). Most of these infections were asymptomatic. Risk factors for transmission included increasing number of units of blood transfused, use of fresh blood, and use of seropositive blood (288). After 1972, transfusions became less common and involved little or no fresh whole blood, especially in cardiac surgery. Concomitant CMV post-transfusion infections have decreased to 1.2% to 17% (288). In contrast to fresh whole blood, leukocytes containing CMV (monocytes and polymorphonuclear leukocytes) survive storage poorly at 2°C to 6°C (288). This observation correlates with the observed reduction of CMV post-transfusion infection rates; studies have shown that 86% of patients infected with CMV after transfusion had received fresh whole blood, compared with 11% of uninfected patients (295,296). Dworkin et al. (297) demonstrated a greater likelihood of isolation of CMV from donor blood during the first 5 days after collection, with infrequent isolation thereafter. In addition to fresh whole blood, granulocyte transfusions have been associated with an especially high risk of CMV infection in compromised hosts (298, 299and300). The difficulty with isolating CMV from donor leukocytes may be a reflection of the small number of leukocytes infected or the need for a posttransfusion host-versus-graft reaction to reactivate the virus (288).
The reduction in overall CMV-related transfusion infections since 1972 also suggests that only a subset of CMV-infected donors (most of whom are seropositive) can transmit infection via donated blood (288,301). This observation is paralleled by the infrequency with which CMV can be isolated from donor blood (288). The receipt of blood from a CMV-seropositive donor significantly correlates with the infrequent residual post-transfusion CMV infections observed since 1972 (302); this has been very well defined in transfused neonates (303). An increased frequency of transmission also correlates with receipt of blood from a donor positive for CMV-IgM antibody (302,304). The pathogenesis and presentation of CMV infection after transfusion involves several factors: the volume of blood transfused, activation of leukocytes harboring CMV, and survival of donor cells in the recipient to allow CMV replication (304).
CMV infection can be diagnosed by direct examination of tissues or exfoliated cells for intranuclear inclusions; however, this lacks sensitivity for active infection (305,306). Isolation of CMV in cell culture from blood (leukocytes) is more sensitive and specific for active infection but labor intensive (1-4 weeks for positivity). The development of fluorescein-labeled monoclonal antibodies for “immediate-early” and “early” antigen detection permits the diagnosis of CMV infection in cell culture within 24 to 48 hours (307, 308, 309and310). Such antibodies can also be used directly to stain biopsied tissue (307,311,312). CMV DNA probes with hybridization and electron microscopy of tissues and leukocytes are limited to research applications generally and have been reported to have lower sensitivity (288,294). “Nested” quantitative PCR has been applied to various body fluids, including cerebrospinal fluid and blood, for the diagnosis of active CMV infection in HIV-1 infected patients (289). This has not been applied to the blood donor setting.
Only gold members can continue reading. Log In or Register to continue
Jun 22, 2016 | Posted by drzezo in GENERAL & FAMILY MEDICINE | Comments Off on Healthcare-Associated Infections Following Transfusion of Blood and Blood Products