7 Haematological disease
Approach to the patient
Clinical features
Investigations
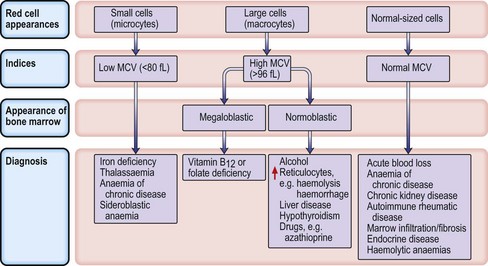
Microcytic anaemias
Iron deficiency anaemia
Investigations
Management
The thalassaemias
α-Thalassaemia
α-Thalassaemia is caused by deletions or mutations in one or more of the four α-globin genes (two genes on each copy of chromosome 16), leading to an excess of β-globin chains. Deletion of one gene results in silent α-thalassaemia, which is of no clinical significance and the blood picture is usually normal. Deletion of two genes results in α-thalassaemia trait (microcytosis with or without mild anaemia but with normal serum iron and ferritin). Haemoglobin H (HbH) disease is caused by a deletion of three α-globin genes, and is characterized by a moderate haemolytic anaemia (Hb 7–10 g/dL) and splenomegaly. Regular transfusion is rarely required, but increased haemolysis may occur with oxidant drugs, as in G6PD deficiency (p. 216), and folic acid should be administered throughout pregnancy. If all four α-globin genes are deleted (Hb Barts), there is a complete absence of normal Hb production, which is incompatible with life outside the uterus (hydrops fetalis).
β-Thalassaemia
• Management of thalassaemia major
Anaemia of chronic disease
A normochromic normocytic anaemia commonly occurs in association with chronic inflammatory and infective conditions (e.g. rheumatoid arthritis, malignancy, TB). The exact pathogenesis is complicated and contributing factors include a cytokine-mediated failure of iron utilization during erythropoiesis and high levels of hepcidin, which destroy ferroportin, limiting iron absorption in the intestinal cell. Red cell survival is decreased and there is an inappropriately low erythropoietin response for the level of anaemia. The MCV can be normal, or low in longstanding disease (resembling iron deficiency). The serum ferritin is usually normal or raised because of the inflammatory process. Iron is present in the bone marrow but not in developing erythroblasts.
Treatment
Address the underlying disorder, e.g. treat infection with antibiotics. Red cell transfusions may be necessary for symptomatic anaemia. Patients do not respond to oral iron, but coexisting iron deficiency should be identified and treated with parenteral iron (p. 202). Recombinant erythropoietin therapy at relatively high doses, e.g. 150–300 U/kg SC three times per week, is used, e.g. in rheumatoid arthritis, or intravenously in chronic kidney disease. Pegylated (PEG) erythropoietin is an alternative, administered once every 2 weeks.
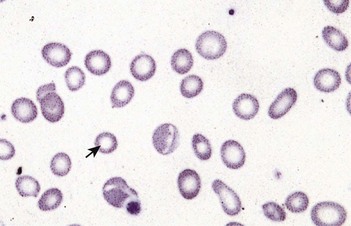
Macrocytic anaemias
Megaloblastic anaemia
Impaired DNA synthesis results in morphological abnormalities of blood cell precursors, characterized by delayed nuclear maturation (nuclear-cytoplasmic asynchrony). All haematopoietic cell lines (and other rapidly dividing cells) are affected, and in severe cases there may be pancytopenia (i.e. anaemia, leucopenia and thrombocytopenia). Causes are predominantly those leading to B12 and folate deficiencies, although drugs (azathioprine, hydroxycarbamide, zidovudine), myelodysplasia and rare enzyme deficiencies affecting DNA synthesis (e.g. orotic aciduria) can also cause macrocytosis and anaemia.
Investigations
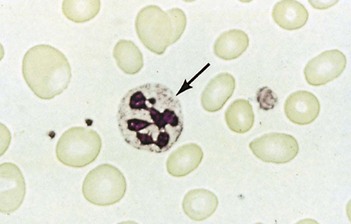
• Other investigations
Treatment
Aplastic anaemia
Treatment
Myelodysplastic syndromes
Investigations
Treatment
Haemolytic anaemias
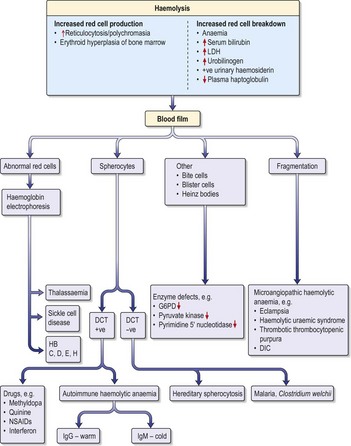
Inherited haemolytic anaemias
Hereditary spherocytosis
Hereditary spherocytosis (HS) is the commonest inherited cause of chronic haemolytic anaemia in Northern Europeans (1 in 5000 births). Inheritance is usually autosomal dominant but 25% of cases are due to spontaneous mutations. The red cells are poorly deformable due to abnormalities in the red cell cytoskeleton, so they become trapped in splenic sinusoids where they are destroyed. HS can present at any age (neonatal jaundice is common) and the clinical features are highly variable. Many patients are asymptomatic but features of chronic compensated haemolysis may be present, e.g. pigment gallstones, mild jaundice and splenomegaly. Aplastic crises sometimes occur after viral infections, e.g. erythrovirus B19. Investigations show anaemia and spherocytes on blood film, with evidence of haemolysis (see above).
• Treatment
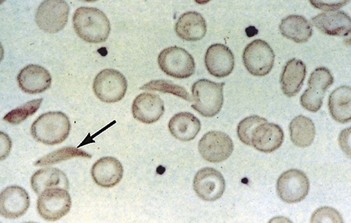
Sickle cell anaemia (HbSS)
Investigations
(See also demonstration of haemolysis, Fig. 7.4).
Clinical syndromes and management
• Analgesia
• Acute chest syndrome (Box 7.2)
Box 7.2
(Adapted from Rees DC et al. 2003.)
Management of acute painful crisis in opioid-naïve adults with sickle cell disease. Higher doses may be required for patients who have previously received opioids.
• Management of acute chest crises
Glucose-6-phosphate dehydrogenase (G6PD) deficiency
Clinical and laboratory features
Rapid intravascular haemolysis with symptomatic anaemia, jaundice and haemoglobinuria occurs 1–3 days after ingestion of certain drugs (Box 7.3). Infection can also precipitate haemolysis but the anaemia is usually mild. Favism (severe haemolysis after ingestion of fava beans) occurs with the Mediterranean variant and can be rapidly fatal. Chronic non-spherocytic haemolytic anaemia (in the absence of a precipitating cause) and neonatal jaundice are also seen in some types of G6PD deficiency.



