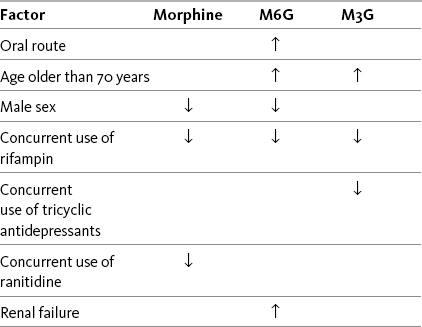
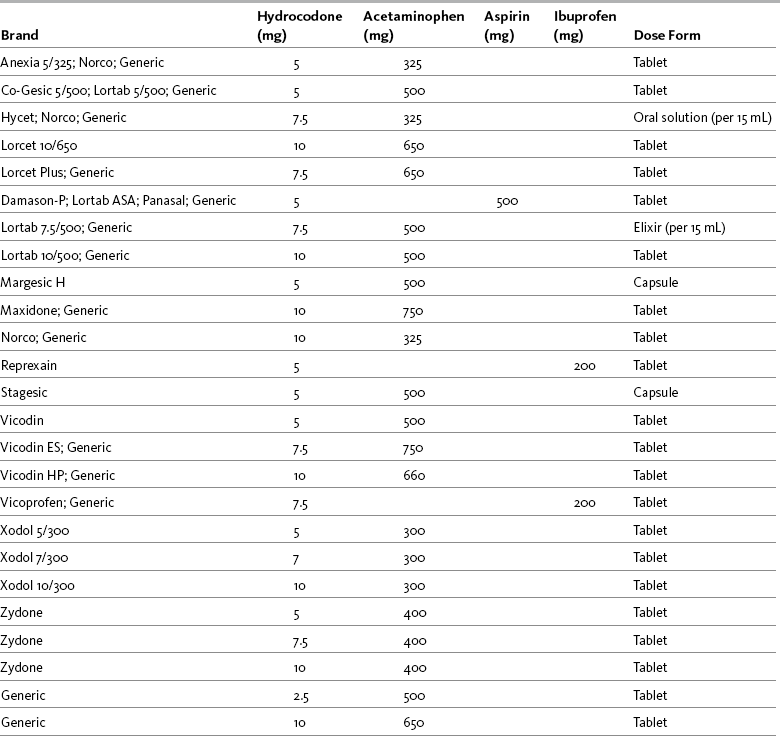
Chapter 13 Goals are stated as simply as possible and shared among the patient, family, and caregivers. Goals ordinarily include the patient’s desired pain rating and the activities that accompany this pain level. For example, a postoperative patient may identify a comfort-function goal of 3/10 to enable regular use of the incentive spirometer and may state that the mild sedation accompanying this level of analgesia is acceptable. A patient with persistent pain may identify a comfort-function goal of 2/10 as necessary for engaging in employment and may state that sedation is not compatible with this goal (see Section II). Many factors are considered when determining the appropriate opioid analgesic for the patient with pain (Box 13-1). These include the unique characteristics of the various opioids and patient characteristics, such as pain intensity, patient age, coexisting disease, current drug regimen and potential drug interactions, prior treatment outcomes, and patient preference. Because pain has multiple underlying mechanisms and is a multifaceted phenomenon, the use of a multimodal approach to managing all types of pain should be the rule, rather than the exception (Argoff, Albrecht, Irving, et al., 2009; Kehlet, Jensen, Woolf, 2006; Kehlet, Wilmore, 2008) (see Section I and Chapter 12). A sound treatment plan relies on the selection of appropriate analgesics from the opioid, nonopioid, and adjuvant analgesic groups. As discussed previously, the mu agonist opioid analgesics are capable of managing all pain intensities and are effective for many different painful conditions. They are the most common analgesics used to manage moderate to severe nociceptive pain, but they have also been shown to be effective for some neuropathic pain (Dworkin, Barbano, Tyring, et al., 2009; Dworkin, O’Connor, Backonja, et al., 2007; Eisenberg, McNicol, Carr, 2006; Gimbel, Richards, Portenoy, 2003; Maier, Hildebrandt, Klinger, et al., 2002; Watson, Moulin, Watt-Watson, et al., 2003). Mu agonists are also recommended for the management of breakthrough pain. See Table 13-1 for a summary of information on selected mu opioid analgesics. For more detail about the characteristics of the various opioid analgesics as they relate to route of administration, see Chapter 14. The equianalgesic chart in Table 16-1 on pp. 444-446 contains dosing and pharmacokinetic information on the various opioid analgesics. Table 13-1 Characteristics of Selected Mu Opioid Agonist Drugs1 1See Table 16-1 on pp. 444-446, for dosing and pharmacokinetic information. 2These are the ratios used in clinical practice. From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, pp. 326-327, St. Louis, Mosby. Data from American Society of Health System Pharmacists. Available at http://www.ashp.org/import/news/HealthSystemPharmacyNews/newsarticle.aspx?id=3037. Accessed December 9, 2009. Barkin, R. L., Barkin, S. J., & Barkin, D. S. (2006). Propoxyphene (dextropropoxyphene), A critical review of a weak opioid analgesic that should remain in antiquity. Am J Ther, 13(6), 534-542; Burnham, R., McNeil, S., Hegedus, C., et al. (2006). Fibrous myopathy as a complication of repeated intramuscular injection for chronic headache. Pain Res Manage, 11(4), 249-252; Chamberlin, K. W., Cottle, M., Neville, R., et al. (2007). Oral oxymorphone for pain management. Ann Pharmacother, 41(7), 1144-1152; Coda, B. A. (2006). Opioids. In P. G. Barash, B. F. Cullen, & R. K. Stoelting (Eds.), Clinical anesthesia, ed 5, Philadelphia, Lippincott, Williams & Wilkins; Dale, O., Hjortkjær, R., & Kharasch, E. D. (2002). Nasal administration of opioids for pain management in adults. Acta Anaesthesiol Scand, 46(7), 759-770; Davis, M. P., Varga, J., Dickerson, D., et al. (2003). Normal-release and controlled-release oxycodone: pharmacokinetics, pharmacodynamics, and controversy. Support Care Cancer, 11(2), 84-92; De Pinto, M., Dunbar, P. J., & Edwards, W.T. (2006). Pain management. Anesthesiology Clin N Am, 24(1), 19-37; Du Pen, S., Du Pen, A., & Hillyer, J. (2006). Intrathecal hydromorphone for intractable nonmalignant pain: a retrospective study. Pain Med, 7(1), 10-15; Fick, D. M., Cooper, J. W., Wade, W. E., et al. (2003). Updating the Beers criteria for potentially inappropriate medication use in older adults: Results of a US consensus panel of experts. Arch Intern Med, 163(22), 2716-2724; Fong, H. K., Sands, L. P., & Leung, J. M. (2006). The role of postoperative analgesia in delirium and cognitive decline in elderly patients: A systematic review. Anesth Analg, 102(4), 1255-1266; Fukuda, K. (2005). Intravenous opioid anesthetics. (2005). In R. D. Miller (Ed.), Miller’s anesthesia, ed 6, St. Louis, Churchill Livingstone; Furlan, A. D., Sandoval, J. A., Mailis-Gagnon, A., et al. (2006). Opioids for chronic noncancer Pain a meta-analysis of effectiveness and side effects. Can Med Assoc J, 174(11), 1589-1594; Gupta, S., & Sathyan, G. (2007). Providing constant analgesia with OROS hydromorphone. J Pain Symptom Manage, 33(2S), S19-S24; Gutstein, H., & Akil, H. (2006). Opioid analgesics. In L. L. Brunton (Ed.), Goodman & Gilman’s the pharmacological basis of therapeutics, ed 11, New York, McGraw-Hill; Hagen, N. A., & Babul, N. (1997). Comparative clinical efficacy and safety of a novel controlled-release oxycodone formulation and controlled-release hydromorphone in the treatment of cancer pain. Cancer, 79, 1428-1437; Hale, M. E., Ahdieh, H., Ma, T., et al. (2007). Efficacy and safety of OPANA ER (oxymorphone extended release) for relief of moderate to severe chronic low back pain in opioid-experienced patients: A 12-week, randomized, double-blind, placebo-controlled study. J Pain, 8(2), 175-184; Hanks, G., Cherny, N. I., & Fallon, M. (2004). Opioid analgesics. In D. Doyle, G. Hanks, N. I. Cherny (Eds.), Oxford textbook of palliative medicine, ed 3, New York, Oxford University Press; Kalso, E. (2005). Oxycodone. J Pain Symptom Manage, 29(Suppl 5), S47-S56; Kumar, M. G., & Lin, S. (2007). Hydromorphone in the management of cancer-related pain: An update on routes of administration and dosage forms. J Pharm Sci, 10(4), 504-518; Latta, K. S., Ginsberg, B., & Barkin, R. L. (2002). Meperidine: A critical review. Am J Ther, 9(1), 53-68; Lugo, R. A., & Kern, S. E. (2004). The pharmacokinetics of oxycodone. J Pain Palliat Care Pharmacother, 18(4), 17-30; McIlwain, H., & Ahdieh, H. (2005). Safety, tolerability, and effectiveness of oxymorphone extended release for moderate to severe osteoarthritis pain. A one year study. Am J Therap, 12(2), 105-112; Miller, M. G., McCarthy, N., O’Boyle, C. A., et al. (1999). Continuous subcutaneous infusion of morphine vs. hydromorphone: A controlled trial. J Pain Symptom Manage, 18(1), 9-16; Mitchell, A., van Zanten, S. V., Inglis, K., et al. (2008). A randomized controlled trial comparing acetaminophen plus ibuprofen versus acetaminophen plus codeine plus caffeine after outpatient general surgery. J Am Coll Surg, 206(3), 472-479; Murray, A., & Hagen, N. A. Hydromorphone. (2005). J Pain Symptom Manage, 29(Suppl 5), S57-66; Prommer, E. (2006). Oxymorphone: A review. Support Care Cancer, 14(2), 109-115; Prommer, E. (2007). Levorphanol: The forgotten opioid. Support Care Cancer, 15, 259-264; Prommer, E. E. (2007). Levorphanol revisited. J Palliat Med, 10(6), 1228-1230; Quigley, C. (2002). Hydromorphone for acute and chronic pain. Cochrane Database of Systematic Reviews, issue 1. Art. No.: CD003447. DOI: 10.1002/14651858.CD003447; Quigley, C., & Wiffen, P. (2003). A systematic review of hydromorphone in acute and chronic pain. J Pain Symptom Manage, 5(2), 169-178; Riley, J., Eisenberg, E., Müller-Schwefe, G., et al. (2008). Oxycodone: A review of its use in the management of pain. Curr Med Res Opin, 24(1), 175-192; Sarhill, N., Walsh, D., & Nelson, K. A. (2001). Hydromorphone: Pharmacology and clinical applications in cancer patients. Support Care Cancer, 9(2), 84-96; Susce, M. T., Murray-Carmichael, E., & de Leon, J. (2006). Response to hydrocodone, codeine and oxycodone in a CYP2D6 poor metabolizer. Prog Neuropsychopharmacol Biol Psychiatry, 30(7), 1356-1358; United States Food and Drug Administration. Available at http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AnestheticAndLifeSupportDrugsAdvisoryCommittee/UCM120095.pdf. Accessed December 10, 2009. Wright, A. W., Mather, L. E., & Smith, M. T. (2001). Hydromorphone-3-glucuronide: A more potent neuro-excitant than its structural analogue, morphine-3-glucuronide. Life Sci, 69(4), 409-420. Pasero C, McCaffery M. May be duplicated for use in clinical practice. Morphine has been administered by several routes of administration: oral, intranasal, intrapulmonary, rectal, IV, SC, IM, intraspinal (epidural and intrathecal), intraarticular, vaginal, sublingual/buccal, and topical (Christensen, Cohen, Mermelstein, et al., 2008; Donnelly, Davis, Walsh, et al., 2002; Hanks, Cherny, Fallon, 2004; Lavelle, Lavelle, Lavelle, 2007; Stoker, Reber, Waltzman, et al., 2008). Poor lipid solubility precludes transdermal absorption and also complicates reliable delivery through mucous membranes, such as the sublingual/buccal route (Donnelly, Davis, Walsh, et al., 2002; Reisfield, Wilson, 2007). The evidence for vaginal administration of morphine is limited to case reports (Ostrop, Lamb, Reid, 1998). Topical application of morphine is reported for painful wounds, in which case it is presumed to have a primary local action; there is a dearth of systematic research on this use, and it should not be considered an approach for systemic analgesic therapy (Paice, Von Roenn, Hudgins, et al., 2008; Zeppetella, Porzio, Aielli, 2007). Nebulized morphine has been used for dyspnea; case reports suggest a favorable local action exists, but the data overall are mixed, and there are no research reports describing this route as a means to provide systemic analgesia (see Chapters 14 and 20). Morphine’s effectiveness as an analgesic, therefore, is established for specific systemic routes of administration—oral and parenteral—and intraspinal routes. Of the parenteral routes, IM administration is not recommended for morphine or for any other drug because of the painful injection and unreliable absorption (APS, 2003) (see Chapter 14). Short-term and long-term parenteral use can be easily accomplished with the IV or SC routes. The oral formulations of morphine are available in liquids, tablets, and capsules and in both short-acting and modified-release preparations. (See Chapter 14 for a detailed discussion of oral morphine formulations.) Morphine is metabolized primarily in the liver. It has two main metabolites, morphine-3-glucuronide (M3G) and morphine-6-glucuronide (M6G) (Gutstein, Akil, 2006). M3G is the primary metabolite of morphine, but it is not active at the opioid receptor and does not produce analgesia (South, Smith, 2001; Andersen, Christrup, Sjogren, 2003); M6G is active at the opioid receptor and produces analgesia (Dahan, van Dorp, Smith, et al., 2008; Smith, Binning, Dahan, 2009; Vaughn, Connor, 2003). Both metabolites have been implicated in morphine toxicity in animals and in patients with advanced disease (Morita, Tei, Tsunoda, et al., 2002), but the studies in humans have not established a clear association (Andersen, Christrup, Sjøgren, 2003). The mechanism by which toxicity, which is evidenced most often by delirium or myoclonus, occurs has not been fully described, and it has been noted that these symptoms are also seen with other opioids (Harris, 2008; Okon, George, 2008) (see Chapter 19 for treatment). Renal insufficiency presumably increases the risk of morphine toxicity because both the parent compound and its major metabolites are renally excreted (Dean, 2004) and there is a direct correlation between creatinine clearance and morphine, M6G, and M3G serum levels. Administration of M3G directly into the CNS has been shown to produce neuroexcitability and anti-analgesic effects in animals (Sharke, Geisslinger, Lotsch, 2005). This metabolite has been implicated as the cause of the neuroexcitability noted in some patients who receive large doses of morphine on a long-term basis (Inturrisi, 2002). Some have suggested that opioid-induced hyperalgesia (see Chapter 11) may be due in part to M3G activity (Hemstapat, Monteith, Smith, et al., 2003; South, Smith, 2001); however, further research in humans is needed to draw firm conclusions (Andersen, Christrup, Sjogren, 2003; Sharke, Geisslinger, Lotsch, 2005). M6G is thought to produce at least some (e.g., 10%) of the analgesic effect of a dose of morphine, but potency and effectiveness studies in humans have produced mixed results (Andersen, Christup, Sjogren, 2003; Smith, South, 2001; Wittwer, Kern, 2006). M6G is absorbed and eliminated from the CNS more slowly than morphine, which may account for the observed increase in potency with long-term morphine administration (Donnelly, Davis, Walsh, et al., 2002; Smith, South, 2001). It may be that the adverse effect profile of M6G is better than that of morphine (Donnelly, Davis, Walsh, et al., 2002). Specific adverse effects that have been investigated include respiratory depression, sedation, nausea and vomiting, hyperalgesia, and myoclonus. A randomized study of 170 patients with moderate to severe postoperative pain demonstrated that M6G produced long-lasting, dose-related analgesia with minimal cardiorespiratory or opioid-like adverse effects (Smith, Binning, Dahan, 2009). In another study (N = 100), M6G was compared with morphine and was found to produce less sedation and respiratory depression, and to have a slower initial onset of effect, with no significant difference in mean pain intensity between groups at 24 hours, but higher pain intensities at 30 minutes and 1 hour after M6G administration (Hanna, Elliott, Fung, 2005). The various factors that influence blood levels of morphine, M6G, and M3G are listed in Table 13-2. Table 13-2 Factors That Influence Blood Levels of Morphine, M6G, and M3G From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 329, St. Louis, Mosby. Data from Buxton, I. L. O. (2006). Pharmacokinetics and pharmacodynamics. The dynamics of drug absorption, distribution, action, and elimination. In L. L. Brunton, J. S. Lazo, & K. L. Parker (Eds.), Goodman & Gilman’s the pharmacological basis of therapeutics, ed 11, New York, McGraw-Hill; Gutstein, H., & Akil, H. (2006). Opioid analgesics. In L. L. Brunton (Ed.), Goodman & Gilman’s the pharmacological basis of therapeutics, ed 11, New York, McGraw-Hill; Portenoy, R. K., & Kanner, R. M. (Eds.). (1996). Pain management: Theory and practice, Philadelphia, FA Davis; Sharke, C., Geisslinger, G., & Lotsch, J. (2005). Is morphine-3-glucuronide of therapeutic relevance? Pain, 116(3), 177-180. Pasero C, McCaffery M. May be duplicated for use in clinical practice. Morphine is hydrophilic (soluble in aqueous solution), which contributes to its slow onset and long duration of action compared with the more lipophilic (soluble in fatty tissue) opioid drugs, such as fentanyl and sufentanil. This is not relevant after steady state is reached during continuous dosing but may be important when intermittent boluses are used systemically or intraspinally (see Chapters 15 through 17). The longer time that it takes morphine to reach its analgesic site of action must be considered when determining how quickly to administer doses during titration (IV); adequate time must be allowed to assess response to one dose before administration of another (Lotsch, Dudziak, Freynhagen, et al., 2006). Morphine has a short half-life of 2 to 4 hours; the half-life of M6G is somewhat longer (Andersen, Christup, Sjøgren, 2003). It is estimated that approximately 20% to 30% of the given dose of oral morphine is available for therapeutic effect because of first-pass effect (De Pinto, Dunbar, Edwards, 2006; Gutstein, Akil, 2006) (see Chapter 11). This is why the recommended dose of morphine by the oral route is higher than that by the parenteral route (APS, 2003) (see Table 16-1 on pp. 444-446). Combination preparations that include codeine are not appropriate for moderate to severe or escalating pain because of the dosing limitations inherent in the nonopioid constituent. The ceiling on the maximum safe daily doses of acetaminophen (4000 mg) and aspirin (4000 mg) limits dose increases for inadequate pain control (see Section III). In addition, aspirin is contraindicated for patients with a number of underlying conditions, such as those with a bleeding disorder or history of asthma. See Patient Education Form VI-2 (pp. 547-548) on codeine with acetaminophen at the end of Section IV. The IM route has been used to administer codeine, but absorption is unreliable and is associated with a five-fold variation in peak blood level; the peak occurs approximately 30 to 60 minutes after IM administration. Nine-fold differences in minimum effective analgesic concentration have been found by this route, and late respiratory depression can occur. These properties make the IM route unfavorable for use in postoperative pain management (Cousins, Umedaly, 1996). It has also long been regarded as inappropriate for IV administration, with low doses of IV morphine recommended instead (Semple, Macintyre, Hooper, 1993) (see Table 13-1).The dose ratio for total analgesic effect between IM and oral codeine is 0.6:1, and a comparison between parenteral codeine and oxycodone found an equianalgesic dose ratio of 10:1; however, its relative potency varies with the extent to which it is converted to its active metabolite (Knotkova, Fine, Portenoy, 2009). Codeine is a prodrug and is approximately 60% bioavailable orally (as compared, for example, with morphine, which has oral bioavailability of 20% to 30%). This is because codeine, like levorphanol, oxycodone, and methadone, undergoes less first-pass metabolism than morphine (Gutstein, Akil, 2006) (see Chapter 11). Once absorbed, 10% of codeine is metabolized in the liver to morphine, its active form (Somogyi, Barratt, Coller, 2007), which probably provides the bulk of its analgesic effect. However, as with other opioids, extremely wide variations exist between individuals in terms of absorption and analgesic requirements of codeine. The metabolism of codeine to morphine depends on the presence of the enzyme cytochrome P450 2D6 (Fine, Portenoy, 2007) (see Chapter 11 for a detailed discussion of this enzyme system). There is population variation in phenotype of cytochrome P450 2D6, distinguishing patients intermediate, extensive (or rapid), ultra-rapid metabolizers, or poor metabolizers. Extensive metabolizers are the norm and are able to perform catalyzed biotransformation of codeine (and other drugs). Approximately 10% of Caucasians and varying frequencies in other ethnic groups are poor metabolizers. These individuals have a very limited ability to convert codeine to morphine, and as a consequence, are relatively less responsive to codeine’s analgesic effect (Palmer, Giesecke, Body, et al., 2005). In contrast, ultra-rapid metabolizers, who biotransform codeine more rapidly or more completely and most of the population, may have an exaggerated (i.e., toxic) response to codeine (Voronov, Przybylo, Jagannathan, 2007; United States Food and Drug Administration [U.S. FDA], 2007a; Palmer, Giesecke, Body, et al., 2005). The impact of the biotransformation via P450 2D6 can vary according to circumstances. Although biotransformation at this isoenzyme occurs at the same rate in neonates and adults, neonates can develop toxicity from codeine because the clearance of morphine is relatively reduced and it can accumulate in the blood. In 2007, the U.S. FDA issued a warning that nursing mothers who are ultra-rapid metabolizers of codeine can transfer sufficient morphine to their breast-feeding infants to cause life-threatening or fatal adverse effects (U.S. FDA, 2007a) (see Chapter 20 for more on opioid use during breast- feeding). In adults, the efficiency of the enzyme can be affected by certain drugs, leading to changes in the production of morphine from codeine. Drugs such as paroxetine (Paxil) and fluoxetine (Prozac), for example, may inhibit cytochrome P450 2D6, and therefore, could potentially interfere with the metabolism of codeine. Case reports suggest that hydrocodone may be effective in CPY2D6 poor metabolizers for whom codeine was ineffective (Susce, Murray-Carmichael, de Leon, 2006). The incidence of moderate-to-severe postoperative pain after craniotomy is common, and the surgical procedure is associated with the development of persistent postsurgical pain (Gottschalk, 2009). Pain management is complicated by concerns about opioid-related adverse effects, such as sedation, miosis, nausea, and vomiting, in this patient population. Codeine has been used for the treatment of postcraniotomy and other types of neurosurgical pain for decades, but its unpredictable absorption, variability in demethylation, and high incidence of nausea and sedation at effective doses make it a particularly poor choice in this population (Roberts, 2004). A review of randomized controlled trials revealed two studies that showed more consistent pain control with morphine compared with codeine and no differences in respiratory depression, sedation, pupillary size, and cardiovascular (CV)effects in patients following craniotomy (Nemergut, Durieux, Missaghi, et al., 2007). A study comparing IV morphine PCA, IV tramadol PCA, and IM codeine in postcraniotomy patients found that morphine produced significantly better analgesia with less vomiting than the other two drugs (Sudheer, Logan, Terblanche, et al., 2007). Roberts (2004) points out that patients are admitted to the intensive care setting where close monitoring is standard following craniotomy. This and thoughtful titration to minimize adverse effects help to ensure the safety of morphine in this population (see also discussion of remifentanil later in this chapter and Chapter 26 for discussion of gabapentin as a component of a multimodal analgesic regimen for craniotomy pain). Fentanyl is the prototype in a subset of mu agonists which includes sufentanil, alfentanil, and remifentanil (see separate discussions for the latter three). All of these drugs are characterized by high potency and high lipophilicity (fat solubility). When administered parenterally to the opioid-naïve patient, the effects are characterized by rapid onset and short duration of action. The injectable formulations of these drugs are administered via the IV, epidural, and intrathecal routes and typically are used for acute pain in the perioperative and procedural settings, often in conjunction with anesthetic or sedating agents; they are the most commonly used opioids in anesthesia (Coda, 2006). They also may be administered transmucosally, e.g., by the buccal, sublingual, or intranasal routes (see Chapter 14). As a class, fentanyl and comparable drugs are versatile, although none are commercially available in an oral or rectal formulation. There are pharmacologic and cost differences that are considered in drug selection for each therapeutic application. With rapid IV administration of high doses, these drugs can produce chest wall rigidity and subsequent difficult ventilation (Lalley, 2005; Fukuda, 2005); this is a concern when fentanyl is used for intraoperative anesthesia (see Chapter 16 for more on speed of injection). Likely related is cough, which has been reported as a complication of both fentanyl and sufentanil (Agarwal, Gautam, Nath, et al., 2007); this effect has been successfully suppressed with IV lidocaine 0.5 mg/kg (Pandey, Raza, Ranjan, et al., 2005). The lipophilicity and potency of fentanyl makes it an excellent candidate for transdermal and oral transmucosal formulation. The fentanyl transdermal patch is commonly used in long-term pain treatment (see Chapter 14). Oral transmucosal fentanyl formulations are used in the treatment of breakthrough pain: oral transmucosal fentanyl and buccal fentanyl; a buccal patch was recently approved in the United States, and a sublingual formulation and an intranasal formulation are available in some other countries (see Chapter 14 for oral transmucosal formulations). Given these kinetics, the half-life of fentanyl varies in the literature depending on whether the study that yielded the value measured the decline in concentration in a steady-state situation or not. Although it has been reported that fentanyl has a terminal half-life of approximately 3 to 4 hours, it is much longer—four to five times longer—after steady state has been approached (Dershwitz, Landow, Joshi-Ryzewicz, 2003; Liu, Gropper, 2003). After steady state is achieved using transdermal fentanyl, half-life also is affected by continued absorption from the skin depot under the patch; the half-life is therefore even longer, typically over 24 hours (see Table 13-1). When used for pain, fentanyl typically is given either parenterally (by the IV route usually) or intraspinally (see Chapter 15). Its rapid onset and short duration in the non–steady state situation make fentanyl the most commonly used opioid in combination with benzodiazepines for procedural analgesia and sedation. Along with morphine and hydromorphone, fentanyl has become a first-line choice for postoperative pain management via IV PCA in many institutions (Pasero, 2005) (see Chapter 17 for PCA dosing). There have been no randomized controlled trials comparing these drugs. A retrospective analysis of medical records compared adverse effects associated with morphine (N = 93), hydromorphone (N = 89), and fentanyl (N = 72) via postoperative IV PCA and found lower mean rates of nausea, pruritus, urinary retention, and sedation with fentanyl; there were no differences among the opioids in incidence of respiratory depression, headache, agitation, confusion, and hallucinations (Hutchison, Chon, Tucker, et al., 2006). However, well-controlled research is needed to draw conclusions regarding differences. Although fentanyl’s properties of high lipophilicity, rapid onset, and short duration in the non–steady state situation make it an attractive option for pain management in a variety of settings, populations, and conditions, these properties also necessitate careful patient selection, appropriate monitoring, and adherence to the safety warnings that accompany fentanyl products, especially the patches and oral transmucosal products. A number of fatalities have occurred due to improper prescribing and use. See Chapter 14 for specific information on indications, patient selection, administration, monitoring, and precautions for the various fentanyl products. Hydrocodone is available in several proprietary products (e.g., Lortab, Vicodin, Lorcet, Hydrocet, and Norco) and generic preparations, and in several different fixed-dose combinations with acetaminophen, aspirin, and ibuprofen. Most are available in tablet form, while others are available in capsule or liquid form (Table 13-3). Combination drugs containing hydrocodone and a nonopioid drug can provide more effective relief than either drug alone. Ibuprofen combined with a variety of hydrocodone doses was shown to increase the effectiveness of hydrocodone seven-fold in animal research (Kolesnikov, Wilson, Pasternak 2003). See Patient Education Form IV-6 on hydrocodone with acetaminophen on pp. 556-557 at the end of Section IV. Table 13-3 Commercially Available Combinations of Hydrocodone and Nonopioids From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 333, St. Louis, Mosby. Pasero C, McCaffery M. May be duplicated for use in clinical practice. Hydrocodone with acetaminophen is not only the most commonly prescribed analgesic in the United States, it is by far the most commonly prescribed medication in all classes (Lamb, 2008). In 2008, hydrocodone plus acetaminophen was first in the list of the 200 most commonly prescribed drugs (Drug Topics, 2008). It is critical to avoid prescribing or administering amounts that would exceed the daily maximum dose for acetaminophen (4000 mg), aspirin (4000 mg), or ibuprofen (3200 mg) and teach the patient the dangers of exceeding these amounts (see Section III). Hydrocodone has an onset of action of approximately 20 minutes, reaches peak effectiveness by 60 minutes, and has a half-life of 3.8 hours (Gutstein, Akil, 2006). It is metabolized by the cytochrome P450 2D6 (CYP2D6) enzyme. Case reports suggest that hydrocodone may be effective in CPY 2D6 poor metabolizers for whom codeine was ineffective (Susce, Murray-Carmichael, de Leon, 2006). (See Chapter 11 for more on the cytochrome P450 enzyme system and drug-drug interactions.) Oral short-acting hydromorphone is available in 2, 4, and 8 mg tablets and in a 1 mg/mL oral solution. It is approximately 60% bioavailable with an onset of action of 30 minutes via the oral route (Kumar, Lin, 2007) and a duration of approximately 3 to 4 hours; maximum plasma concentrations are reached within 1 hour of dosing (Gupta, Sathyan, 2007). Modified-release formulations of oral hydromorphone are available in Canada and Europe and most recently on the U.S. market (Gupta, Sathyan, 2007) (see Chapter 14 for modified-release hydromorphone). See Patient Education Form IV-3 on short-acting hydromorphone at the end of Section IV. Hydromorphone also has been administered via a variety of other routes. Its greater potency compared with morphine, as well as its availability in concentrated parenteral form (10 mg/mL), has made it attractive for SC administration, especially when high doses are needed. It was found to be comparable to morphine by SC continuous infusion for persistent cancer pain in terminally ill patients (Miller, McCarthy, O’Boyle, et al., 1999). Absorption via the IM route is erratic and not recommended (Golembiewski, 2003). The epidural and intrathecal routes have been utilized for acute and persistent cancer and noncancer pain (DuPen, DuPen, Hillyer, 2006). Hydromorphone given rectally (3 mg suppository) is as effective as by the oral route; it is not, however, absorbed well by the oral mucosa (Kumar, Lin, 2007; Sarhill, Walsh, Nelson, 2001) (see Table 13-1). The equianalgesic dose conversion between morphine and hydromorphone is unclear and likely varies with the length of time a patient has been on one drug or the other (Berdine, Nesbit, 2006; Knotkova, Fine, Portenoy, 2009). Published equianalgesic tables typically show oral hydromorphone to be 5 times more potent than oral morphine. This is the most common ratio used when preparing equianalgesic solutions for PCA administration (e.g., 0.2 mg hydromorphone per 1 mL solution is considered approximately equal to 1 mg morphine per 1 mL solution) (Golembiewski, 2003). However, these data are generally derived from acute pain treatment in opioid-naïve patients or healthy volunteers (APS, 2003). An early study of morphine-hydromorphone equivalence showed that after a week of PCA treatment, the ratio was 3:1 (morphine 10 mg to hydromorphone 3.3 mg) (Dunbar, Chapman, Buckley, et al., 1996). Subsequent research found that when switching from long-term dosing of either oral or parenteral morphine to hydromorphone, the ratio was approximately 5.5:1 (morphine 10 mg to hydromorphone 2 mg) (Lawlor, Turner, Hanson, et al., 1997). However, when switching from hydromorphone to morphine, the ratio was 3.7:1 (morphine 10 mg to hydromorphone 2.7 mg). Still others suggest that an equianalgesic dose conversion of parenteral morphine to hydromorphone for long-term dosing is probably 4:1 (morphine 10 mg to hydromorphone 2.5 mg) (Hanks, Cherny, Fallon, 2004). A systematic review of hydromorphone for various types of pain concluded that there is insufficient evidence to recommend specific ratios of hydromorphone (Quigley, Wiffen, 2003) (see also Knotkova, Fine, Portenoy, 2009). (See Table 16-1). Like methadone, levorphanol (Levo-Dromoran) is considered a second-line drug for cancer pain (Hanks, Cherny, Fallon, 2004). It has not been widely used clinically since the modified-release formulations of morphine and oxycodone became available in the 1990s (McNulty, 2007). Most clinicians are unfamiliar with its pharmacology, which is somewhat different than the more commonly used opioids (McNulty, 2007; Prommer, 2007a; Prommer, 2007b). It is an agonist at both the mu and kappa opioid receptor sites and, like methadone, it also is an N-methyl-d-aspartate (NMDA) antagonist. In addition, it is an inhibitor of serotonin and norepinephrine reuptake (see Section I). Levorphanol is available in the United States orally only in 2 mg tablets, which can make titration difficult. It is no longer available in the United Kingdom or Canada (Hanks, Cherny, Fallon, 2004). (See Table 16-1 on pp. 444-446.) Meperidine (Demerol) was once the most widely used opioid analgesic. In recent years, it has been either removed from or severely restricted on hospital formularies, the result of concerted efforts to improve patient safety during opioid use (Gordon, Jones, Goshman, et al., 2000; Raymo, Camejo, Fudin, 2007) (see the paragraphs that follow). The Beers Criteria of inappropriate medication use in older individuals, originally developed in 1991 (Beers, Ouslander, Rollingher, et al., 1991), described meperidine as having many disadvantages and continues to advise against the use of the drug in older adults (Beers, 1997; Fick, Cooper, Wade, et al., 2003) (Table 13-4). A refinement of the 1996 Medical Expenditure Panel Survey designated the drug as one to “always avoid” in older adults (Zhan, Sangl, Bierman, et al., 2001). Meperidine has some positive attributes, but it continues to be overused (Kornitzer, Manace, Fischberg, 2006) and misused (Hubbard, Wolfe, 2003) because of lack of knowledge about its pharmacology. Numerous misconceptions about meperidine persist (Table 13-5). Table 13-4 Beers Criteria for Inappropriate Medication Use in Older Adults: Selected Analgesics CNS, Central nervous system; CV, cardiovascular; GI, gastrointestinal, From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 336, St. Louis, Mosby. Data from Beers, M. H. (1997). Explicit criteria for determining potentially inappropriate medication use by the elderly. An update. Arch Intern Med, 157(14), 1531-1536; Beers, M. H., Ouslander, J. G., Rollingher, I, et al. (1991). Explicit criteria for determining inappropriate medication use in nursing home residents. UCLA Division of Geriatric Medicine. Arch Intern Med, 151(9), 1825-1832; Fick, D. M., Cooper, J. W., Wade, W. E., et al. (2003). Updating the Beers criteria for potentially inappropriate medication use in older adults: Results of a US consensus panel of experts. Arch Intern Med, 163(22), 2716-2724; Zhan, C., Sangl, J., Bierman, A. S., et al. (2001). Potentially inappropriate medication use in the community-dwelling elderly: Findings from the 1996 Medical Expenditure Panel Survey. JAMA, 286(22), 2823-2829. Pasero C, McCaffery M. May be duplicated for use in clinical practice. Table 13-5 IM, Intramuscular; mg, milligram. From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 337, St. Louis, Mosby. Data from Austin, K. L., Stapleton, J. V., & Mather, L. E. (1980). Relationship between blood meperidine concentrations and analgesic response: A preliminary report. Anesthesiology, 53(6), 460-466; Beaulé, P. E., Smith, M. I., & Nguyen, V. N. (2004). Meperidine-induced seizure after revision hip arthroplasty. J Arthroplasty, 19(4), 516-519; Burnham, R., McNeil, S., Hegedus, C., et al. (2006). Fibrous myopathy as a complication of repeated intramuscular injection for chronic headache. Pain Res Manage, 11(4), 249-252; Coelho, J. C., Senninger, N., Runkel, N., et al. (1986). Effect of analgesic drugs on electromyographic activity of the gastrointestinal tract and sphincter of Oddi and on biliary pressure. Ann Surg, 204(1), 53-58; Fong, H. K., Sands, L. P., & Leung, J. M. (2006). The role of postoperative analgesia in delirium and cognitive decline in elderly patients: A systematic review. Anesth Analg 102(4), 1255-1266; Hubbard, G. P., & Wolfe, K. R. (2003). Meperidine misuse in a patient with sphincter of Oddi dysfunction. Ann Pharmacother, 37(4), 534-537; Kornitzer, B. S., Manace, L. C., Fischberg, D. J., et al. (2006). Prevalence of meperidine use in older surgical patients. Arch Surg, 141(8), 76-81; Latta, K. S., Ginsberg, B., & Barkin, R. L. (2002). Meperidine: A critical review. Am J Ther, 9(1), 53-68; Lee, F., & Cundiff, D. (1998). Meperidine vs morphine in pancreatitis and cholecystitis. Arch Intern Med, 158(21), 2399; Mohta, M., Kumari, N., Tyagi, A., et al. (2009). Tramadol for prevention of postanaesthestic shivering: A randomised double-blind comparison with pethidine. Anaesthesia, 64(2), 141-146; Kranke, P., Eberhart, L. H., Roewer, N., et al. (2004). Single-dose parenteral pharmacological interventions for prevention of postoperative shivering: A quantitative systematic review of randomized controlled trials. Anesth Analg, 99(3), 718-727; Radnay, P. A., Brodman, E., Mankikar, D., et al. (1980). The effect of equianalgesic doses of fentanyl, morphine, meperidine, and pentazocine on common bile duct pressure. Anaesthetist 29, 26-29; Schwarzkopf, K. R. G., Hoff, H., Hartmann, M., et al. (2001). A comparison between meperidine, clonidine and urapidil in the treatment of postanesthetic shivering. Anesth Analg, 92(1), 257-260. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
Guidelines for Opioid Drug Selection
Characteristics of Selected Mu Agonist Opioids
Mu Opioid Agonist Drug
Routes Administered
Comments
Morphine
PO (short-acting and modified-release), SL, R, IV, IM, SC, E, I, IA
Standard for comparison. Multiple routes of administration. Several modified-release formulations available, but they are not therapeutically equivalent. Begin with lower doses in older adults. Active metabolite M6G can accumulate with repeated dosing in renal failure. 20% to 30% oral bioavailability.
Codeine
PO, IM, SC
Limited usefulness. Usually compounded with nonopioid (e.g., Tylenol No. 3). Used orally for mild to moderate pain, but analgesia is inferior to that of ibuprofen. IM has unpredictable absorption and high adverse effect profile; IV route not recommended, SC rarely used, and IM administration of any opioid is discouraged.
Fentanyl
OT, B, IV, IM, TD, E, I, IN
Fast-acting; short half-life (except TD). At steady state, slow elimination from tissues can lead to a prolonged half-life (up to 12 h). On the basis of clinical experience, fentanyl 1 mcg/h transdermally is roughly equivalent to morphine 2 mg/24 h orally2; fentanyl, 100 mcg/h parenterally and transdermally is roughly equivalent to 4 mg/h morphine parenterally.2 Opioid-naïve patients should be started on no more than 25 mcg/h transdermally. Transdermal fentanyl is not appropriate for acute pain management. OTFC and buccal fentanyl are approved for management of breakthrough pain in opioid tolerant individuals.
Hydrocodone
PO
Used for mild to moderate pain; available in nonopioid combination only (e.g., Vicodin, Lortab) (see Table 13-3).
Hydromorphone (Dilaudid)
PO, R, IV, IM, SC, E, I
Useful alternative to morphine. Metabolite may accumulate with long-term, high dose administration. Available in high-potency parenteral formulation (10 mg/mL) useful for SC infusion; 3 mg R roughly equivalent to 650 mg aspirin; oral modified-release formulation available.
Levorphanol (Levo-Dromoran)
PO, IV, IM, SC
Long half-life can lead to accumulation within 2 to 3 days of repetitive dosing.
Meperidine (Demerol)
PO, IV, IM, SC, E, I
No longer recommended for the management of any type of pain because of potential toxicity from accumulation of metabolite, normeperidine. Half-life of normeperidine is approximately 15 to 20 h; NR in older adults or patients with impaired renal function; continuous IV infusion NR. The most appropriate candidates for meperidine use are patients with acute pain who are otherwise healthy with no risk factors and are allergic to or intolerant of other opioids, such as morphine, fentanyl, and hydromorphone, or have demonstrated a more favorable outcome with meperidine than other opioid drugs.
Methadone (Dolophine)
PO, SL, R, IV, SC, IM, E, I
Long half-life can lead to delayed toxicity from accumulation. See text for information on methadone.
Oxycodone (OxyIR, OxyContin)
PO (short-acting and modified-release), IV, IM, R
Used for mild to moderate pain when combined with a nonopioid (e.g., Percocet, Tylox) (see Table 13-9). As single entity, can be used like oral morphine for severe pain. Rectal and parenteral formulation not available in the United States. Oral formulation can be administered rectally.
Oxymorphone (Opana, Opana ER [oral], Numorphan [parenteral, rectal])
PO (short-acting and modified-release) IV, IM, SC, R
Used for moderate to severe pain. Available in 5 mg rectal suppositories.
Propoxyphene (Darvocet, Darvon)
PO
Used in combination with acetaminophen (Darvocet) and aspirin (Darvon Compound). Long half-life. Accumulation of toxic metabolite norpropoxyphene with repetitive dosing. Inappropriate for use in older adults (see Table 13-4).
Morphine
Codeine
Codeine Post-Craniotomy
Fentanyl
Hydrocodone
Hydromorphone
Levorphanol
Meperidine
Drug
Concerns
Severity Rating
Propoxyphene (Darvon, Darvocet, Darvon Compound)
Offers no advantages over other opioids; toxic metabolite; high adverse effect profile.
Low
Meperidine (Demerol)
Offers few if any advantages over other opioids; toxic metabolite that can cause CNS disturbances.
High
Pentazocine (Talwin)
Low analgesic efficacy; high incidence of CNS adverse effects (e.g., hallucinations, delirium).
High
Short-acting benzodiazepines (e.g., Ativan, Restoril, Serax, Xanax)
Increased sensitivity in older adults; dose-related adverse CNS effects.
High
Long-acting benzodiazepines (e.g., Librium, Valium)
Increased sensitivity in older adults; long half-life; dose-related adverse CNS effects; associated with falls and fractures. Short-acting is preferred if a benzodiazepine is needed.
High
Flurazepam (Dalmane)
Very long half-life in older adults (often days); prolonged.
High
Anticholinergics and antihistamines (e.g., Benadryl, Atarax, Vistaril)
Most antihistamines have potent anticholinergic effects. Noncholinergic antihistamines are preferred. Hydroxyzine (Vistaril, Atarax) and diphenhydramine (Benadryl) can cause confusion and sedation. If antihistamine is necessary, use low dose.
High
Amitriptyline (Elavil)
High incidence of anticholinergic and sedative adverse effects; rarely appropriate in older adults.
High
Daily fluoxetine (Prozac)
Long half-life; can produce CNS stimulation, sleep disturbances, and agitation.
High
Clonidine (Catapres)
Can cause orthostatic hypotension and CNS adverse effects.
Low
Orphenadrine (Norflex)
Can cause significant sedation and anticholinergic effects.
High
Muscle relaxants (e.g., Soma, Flexeril, Skelaxin)
Most have anticholinergic effects and are poorly tolerated by older adults. Can cause sedation and muscle weakness, which may contribute to falls.
High
Long-term use of full-dose, longer half-life, nonselective NSAIDs (e.g., Naprosyn, Aleve, Feldene)
Potential GI, renal, CV adverse effects.
High
Ketorolac (Toradol)
High incidence of GI adverse effects in older adults.
High
Indomethacin (Indocin)
High incidence of CNS adverse effects.
High
Misconception
Correction
Meperidine causes less respiratory depression than morphine.
At equianalgesic doses, opioid analgesics produce equal respiratory depression.
Meperidine is less likely than morphine to cause addiction.
The abuse liability for meperidine is at least as high as that for morphine. In other words, people addicted to opioids find morphine and meperidine equally attractive. Several early reports suggested meperidine may be the more addictive of the two.
Meperidine causes less constriction of the sphincter of Oddi and the biliary tract than does morphine.
Both meperidine and morphine cause constriction of the sphincter of Oddi and the biliary tract. Laboratory studies show that morphine may cause more constriction in animals, but this has never been shown to be clinically relevant in humans. In humans, morphine and meperidine caused a rise in bile duct pressure of 52.7% and 61.3%, respectively.
Meperidine is less constipating than morphine.
Meperidine may be less constipating but only when used on a long-term basis, and long-term use is not recommended.
Long-term clinical experience with meperidine proves it is safe and effective.
Meperidine prescribing has declined, but the drug continues to be used despite ample evidence that it has no advantages over other opioids and has toxicities that make it undesirable for almost any use. Historically, therapeutic doses (e.g., 100 mg IM for adults) were seldom used, and studies show that during decades of use, many patients were undertreated for pain. Furthermore, problems may have gone unnoticed because the existence of the metabolite normeperidine was not known and patients were not assessed for signs of neurotoxicity. Meperidine cannot be used safely if pain is treated aggressively.
Meperidine is the only drug effective for treatment of perioperative and postdelivery shivering.
Although low-dose meperidine is widely used to treat perioperative and post-delivery shivering, other drugs are also effective. These include clonidine, ondansetron, and tramadol.
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Guidelines for Opioid Drug Selection
Only gold members can continue reading. Log In or Register to continue
