54 Gout and hyperuricaemia
Epidemiology
Gout is one of the oldest recognised diseases and was identified by the Egyptians in 2460 BC. Hippocrates described it as ‘arthritis of the rich’ due to the association with certain foods and alcohol. Gout affects 1–2% of adults in developed countries, and in recent decades there has been a significant rise in its prevalence and incidence (Zhang et al., 2006). The USA has seen a doubling in the number of cases with the rate of gout increasing to 4.1% in older males. However, unlike the rest of the world, prevalence in the UK appears not to be rising and from 2000 to 2005 remained at 1.4% (Rider and Jordan, 2010). The increasing numbers in many developed countries have been attributed to trends in lifestyle leading to increased risk of gout, for example, obesity, metabolic syndrome, hypertension, alcohol consumption and increased age of the general population. Although the Maori population have a marked genetic predisposition to gout, prior to 1700 they did not experience this inflammatory joint disease. It was changes in diet and lifestyle following European settlement that led to the appearance and increasing prevalence in the country. New Zealand now has probably the highest prevalence in the World with one in eight men affected (Richette and Bardin, 2010). A similar pattern has also been seen in Eastern China, where gout was considered a very rare disease in the 1980s. Changes in diet and lifestyle due to Western influences have seen its prevalence rise to 1.1% in Eastern China in 2008.
In the UK, the presentation of gout in men before the age of 45 years is unusual, but in those over the age of 75 years prevalence is greater than 7% in men and 4% in women (Doherty, 2009; Jordan et al., 2007; Zhang et al., 2006). Gout is predominantly a disease of men with a male to female ratio of 3.6:1. In women, it tends to develop after menopause when levels of oestrogen, a known uricosuric, fall.
Although environmental factors are clearly implicated in the development of gout, studies have shown that inheritance also plays an important role. In recent years, research into the genetic background of gout has identified several renal urate transporters including URAT-1 and GLUT-9 and the genes that encode them, for example, SLC22A12 and SLC2A9, respectively. Polymorphisms in these genes are associated with increased hyperuricaemia and gout (Dalbeth and Merriman, 2009; Doherty, 2009).
Pathophysiology
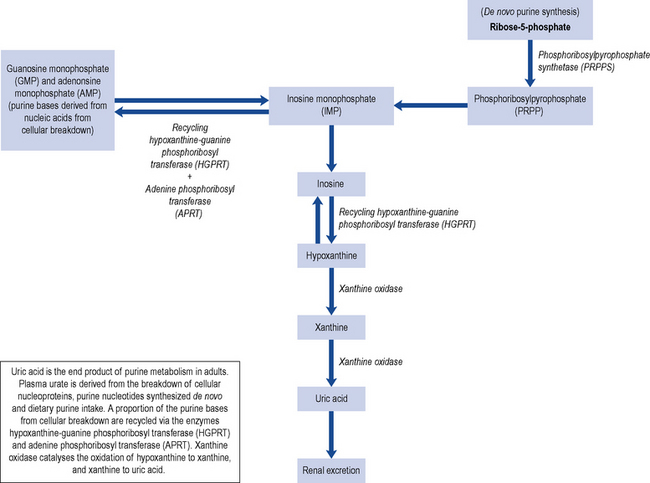
Uric acid is mainly a by-product from the breakdown of cellular nucleoproteins and purine nucleotides synthesised de novo with about a third coming from the breakdown of dietary purine intake (Fig. 54.1). Uric acid is a weak acid with a pKa of 5.75, and at the physiological pH of the extra-cellular compartment 98% of uric acid is in the ionised form of urate. This is mainly present as monosodium urate due to the high concentration of sodium in the extra-cellular compartment. Human beings and higher primates lack the enzyme uricase that degrades uric acid to the highly soluble allantoin resulting in higher concentrations of urate close to the level of solubility. Monosodium urate has a solubility limit of 380 µmol/L; when the concentration exceeds 380 µmol/L, there is a risk of precipitation and the formation of monosodium urate crystals.
Primary gout is not a consequence of an acquired disorder, but is associated with rare inborn errors of metabolism and isolated renal tubular defects in the fractional clearance of uric acid. A rare group of enzyme defects result in an increased de novo purine synthesis such as hypoxanthine-guanine phosphoribosyl transferase deficiency (Lesch-Nyhan syndrome), phosphoribosyl pyrophosphate synthetase super activity, glucose-6 phosphatase deficiency and myogenic hyperuricaemia (Table 54.1).
Table 54.1 Causes of primary and secondary gout
| Primary gout | Secondary gout |
|---|---|
| Idiopathic Rare enzyme deficiencies Hypoxanthine-guanine phosphoribosyl transferase deficiency (HPRT) Phosphoribosyl pyrophosphate synthetase super-activity Ribose-5-phosphate AMP-deaminase deficiency | Increased uric acid production Lymphoproliferative/Myeloproliferative Chronic haemolytic anaemias Secondary polycythemia Severe exfoliative psoriasis Gaucher’s disease Cytotoxic drugs Glucose-6 phosphate deficiency High purine diet overproduction Reduced uric acid secretion Renal failure Hypertension Drugs (diuretics, aspirin, ciclosporin) Lead nephropathy Alcohol Down’s Syndrome Myxoedema Beryllium poisoning |
Secondary gout is the consequence of the use of specific drugs or develops as a consequence of other disorders. Certain diseases are associated with enhanced nucleic acid turnover, for example, myeloproliferative and lymphoproliferative disorders, psoriasis and haemolytic anaemia, and can lead to hyperuricaemia. Renal mechanisms are responsible for the majority of hyperuricaemia in individuals with over production representing less than 10% of patients with gout. The kidney excretes about two-thirds of the uric acid produced daily with the remainder being eliminated via the biliary tract with subsequent conversion to soluble allantoin by colonic bacterial uricase. Approximately 90% of the daily load of urate filtered by the kidneys is re-absorbed. This re-absorption process is mediated by specific anion transporters such as URAT-1 which is located on the apical side of the renal proximal tubular cells and is an important determinant of urate re-absorption (Richette and Bardin, 2010). The URAT-1 transporter is targeted by a number of drugs including benzbromarone, probenecid, losartan and sulphinpyrazone.
Risk factors
Hyperuricaemia is one of the main risk factors for gout and occurs in about 15–20% of the population (Doherty, 2009). Fortunately, only a minority of individuals with increased serum uric acid levels develop gout suggesting the importance of other contributing factors (Box 54.1).
Box 54.1 Risk factors for gout
Co-morbidities, for example, obesity, dyslipidaemia, glucose intolerance, hypertension
Genetics
Common primary gout in men often shows a strong familial predisposition, although the genetic basis for this is not fully understood. A polymorphism of the SLC22A12 gene which encodes for URAT-1 has been associated with under excretion of uric acid and hyperuricaemia in German Caucasians (Graessler et al., 2006). While in a Japanese cohort, another mutation of the SLC22A12 gene has been shown to be protective for the development of gout (Taniguchi et al., 2005).
The recently identified glucose and fructose transporter (GLU9) also acts as a high-capacity urate transporter in the proximal renal tubules (Dalbeth and Merriman, 2009). Polymorphism in the gene which encodes for this transporter (SLC2A9) has been reported to influence serum uric acid levels, and a significant association with self-reported gout has been described (Dalbeth and Merriman, 2009).
Renal disease
Gout is frequently associated with kidney disease, each being a risk factor for the other. Hyperuricaemia is associated with primary kidney disease, but kidney damage may arise secondary to gout as a consequence of the deposition of urate crystals in the interstitium and tubules of the kidney. Historically, gout was associated with significant renal impairment; however, progressive renal failure directly due to gout is now rare and mainly limited to inadequately treated patients with primary purine overproduction associated with purine enzyme defects, rare forms of inherited renal disease, chronic lead intoxication and renal disease as a consequence of uncontrolled disease states associated with gout e.g. hypertension, type 2 diabetes and congestive cardiac failure. Men with gout have a two-fold higher risk of kidney stones than patients without gout (Jordan et al., 2007). The likelihood of stones increases with serum urate concentration, extent of urinary acid secretion and low urine pH.
Co-morbidities
Metabolic syndrome is a multiplex risk factor for atherosclerotic cardiovascular disease that consists of atherogenic dyslipidaemia, raised blood pressure, increased blood glucose, and both prothrombotic and pro-inflammatory states. In the USA, metabolic syndrome is present in 63% of those with gout compared to 25% of those without gout (Choi et al., 2007). Other studies have shown obesity, weight gain and hypertension all to be independent risk factors for the development of gout (Choi et al., 2005).
Diet
Gout has often been associated with a rich lifestyle and excesses in diet. In particular, gout is higher in people who consume large quantities of red meat. There is also an increased risk associated with seafood consumption, but to a lesser extent than with red meat. In contrast, a diet high in purine-rich vegetables does not increase the risk, and the consumption of low-fat dairy products reduces the relative risk of gout with each additional dairy serving. The consumption of soft drinks sweetened with sugar (not diet drinks) has also been linked to an increase in the number of gout cases particularly in USA (Choi and Curham, 2008). The mechanism of action is thought to be an increase in uric acid levels caused by an increase in adenine nucleotide degradation. Vitamin C (ascorbic acid) has been shown to have a modest uricosuric effect (Huang et al., 2005). The consumption of cherries, but no other fruits, has also been shown to decrease uric acid levels.
Alcohol
Increased daily consumption of alcohol is associated with a higher risk of gout. Beer carries the greatest risk, probably due to its high purine content, followed by spirits. However, a moderate consumption of wine is not associated with an increased risk of developing gout (Jordan et al., 2007). The mechanism of action involved is thought to be the metabolism of ethanol to acetyl coenzyme A leading to adenine nucleotide degradation, with resultant increased formation of adenosine monophosphate, a precursor of uric acid. Alcohol also raises lactic acid levels in blood, which inhibits uric acid excretion.
Medication
A number of drugs are associated with increased uric acid levels (Box 54.2). The use of both loop and thiazide diuretics is the most common modifiable risk factor for secondary gout, especially in the elderly. It is thought loop and thiazide diuretics may precipitate an attack via volume depletion and reduced renal tubular secretion of uric acid. Aspirin has a bimodal effect; low doses inhibit uric acid excretion and increase urate levels, while doses greater than 3 g/day are uricosuric.
The prescribing of ciclosporin in organ transplant patients is an independent risk factor for new-onset gout in this group. The proposed mechanism of action is the interaction of ciclosporin with the hOAT10 transporter that mediates urate/glutathione exchange in the kidney (Bahn et al., 2008). Radiotherapy and chemotherapy in patients with neoplastic disorders can cause hyperuricaemia because of increased cell breakdown; to overcome this, prophylactic treatment may be given with allopurinol, commencing 3 days before therapy.
Presentation and diagnosis
The shed crystals are phagocytosed by monocytes and macrophages, activating the NACHT–LRR–PYD-containing protein-3 (NALP3) inflammasome and triggering the release of interleukin-1β (IL-1β) and other cytokines, a subsequent infiltration of neutrophils and the symptoms of an acute attack (Dalbeth and Haskard, 2005). The NALP3 inflammasome (cryopyrin) is a complex of intracellular proteins that is activated on exposure to microbial elements, such as bacterial RNA and toxins. Activation of NALP3 leads to the release of caspase-1, which is required for cleavage of pro-IL-1β to active IL-1β (Richette and Bardin, 2009). IL-1β has been shown to be critically associated with the inflammatory response induced by monosodium urate crystals (Rider and Jordan, 2010).
A third of patients will have normal uric acid concentrations during an acute attack of gout due to increased urinary urate excretion. The most appropriate time to measure serum urate for monitoring purposes is when the attack has completely resolved. The gold standard for the diagnosis of gout is the demonstration of urate crystals in synovial fluid or in a tophus by polarised light microscopy (Zhang et al., 2006). Crystals may be found in fluid aspirated from non-inflamed joints, even in those joints which have not previously experienced an attack. The crystals are large (10–20 μm) and needle shaped with a strong, intense, characteristic light pattern under polarised light. In contrast, the calcium pyrophosphate dehydrate crystals associated with pseudo-gout are small rhomboid crystals of low intensity. Gout and septic arthritis may co-exist and in order to exclude septic arthritis synovial fluid is sent for Gram staining and culture.
Course of disease
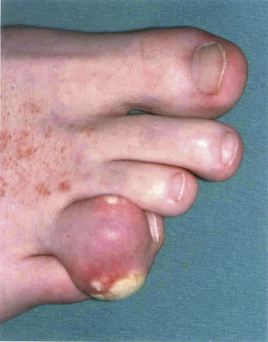
The course of gout follows a number of stages; initially, the patient may be asymptomatic with a raised serum uric acid level (Fig. 54.2). Some patients may only ever experience one attack, but often a second attack occurs within 6–12 months. Subsequent attacks tend to be of longer duration, affect more than one joint and may spread to the upper limbs. Untreated disease can result in chronic tophaceous gout, with persistent low-grade inflammation in a number of joints resulting in joint damage and deformity. The disease is characterised by the presence of tophi (Fig. 54.3), monosodium urate crystals surrounded by chronic mononuclear and giant-cell reactions. Tophi deposition can occur anywhere in the body, but they are commonly seen on the helix of the ear, within and around the toe or finger joints, on the elbow, around the knees or on the Achilles tendons. The skin overlying the tophi may ulcerate and extrude white, chalky material composed of monosodium urate crystals.
Treatment
The management of gout can be split into the rapid resolution of the initial acute attack and long-term measures to prevent future episodes (see Box 54.3).
Box 54.3 Treatment aims in gout
Rapid alleviation of the acute attack
Lower serum uric acid levels to below saturation point
Reduce risk of co-morbidities, for example, cardiovascular disease
Management of an acute attack
Drugs used in the management of an acute attack include NSAIDs, colchicine and corticosteroids. NSAIDs are the recommended first-line agents, but in a number of patients their use is contraindicated and a second-line agent is indicated (Box 54.4). Where the pain is not adequately controlled by treatment, paracetamol and weak opiate analgesics, for example, codeine or dihydrocodeine may be added to the regimen to provide additional relief. Treatment should be continued until the attack is terminated, usually between 1 and 2 weeks. The affected joints should also be rested for 1–2 days and initially treated with ice which has a significant analgesic effect during an acute attack.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


