Glycogen Storage Disease
Monica P. Revelo, MD, PhD
Key Facts
Terminology
Inherited autosomal recessive disorders due to deficiency in lysosomal enzymes resulting in glycogen accumulation that causes cellular dysfunction and progressive damage of liver, heart, and skeletal muscle
Macroscopic Features
Massive cardiomegaly
Marked thickening of ventricular walls
Microscopic Pathology
Marked vacuolar change of myocyte cytoplasm with lacework appearance
Variable degree of interstitial fibrosis
Ancillary Tests
Myocardial glycogen deposits washed out after diastase treatment
EM shows cytoplasmic accumulation of glycogen in myocardium with granular or fibrillary appearance
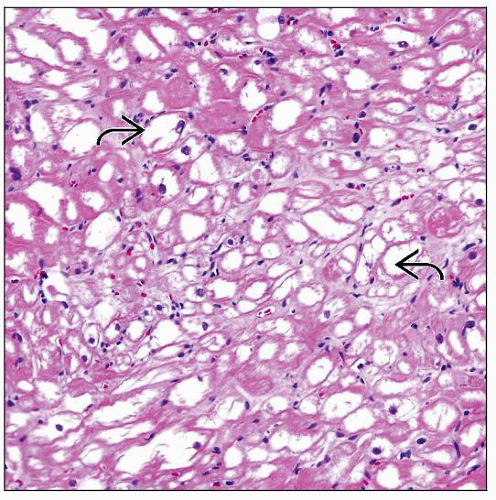 H&E-stained section at low magnification shows myocardium with marked cytoplasm vacuolization of myocytes with empty appearance and paucity of myofibrils  . . |
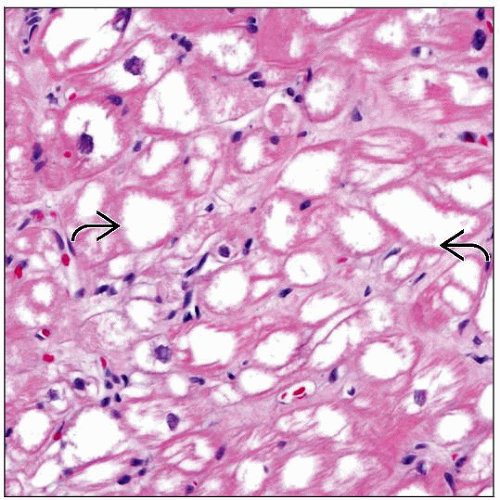 H&E-stained section at higher magnification shows myocytes with lacework appearance, empty-appearing cytoplasm, and loss of myofibrils  . . |
TERMINOLOGY
Definitions
Inherited autosomal recessive disorders due to deficiency in lysosomal enzymes resulting in glycogen accumulation that causes cellular dysfunction and progressive damage of liver, heart, and skeletal muscle
ETIOLOGY/PATHOGENESIS
Inherited Enzyme Deficiency
Glycogen storage disease II (Pompe disease): α-1,4-glucosidase
Glycogen storage disease III (Cori disease): Amylo-1,6-glucosidase
Glycogen storage disease IV (Andersen disease): α-1,4-glucan 6-glycosyl transferase
AMP-activated protein kinase deficiency: γ-2 regulatory subunit of enzyme controlling uptake of glucose
CLINICAL ISSUES
Presentation



