Glomerulonephritis
KEY CONCEPTS
![]() Glomerulonephritis is a collection of glomerular diseases mediated by different immunologic pathogenic mechanisms, resulting in varied clinical presentation and therapeutic outcomes.
Glomerulonephritis is a collection of glomerular diseases mediated by different immunologic pathogenic mechanisms, resulting in varied clinical presentation and therapeutic outcomes.
![]() The signs and symptoms associated with glomerulonephritis can be nephritic in nature, characterized by inflammatory injury, or nephrotic in nature, characterized by proteinuria.
The signs and symptoms associated with glomerulonephritis can be nephritic in nature, characterized by inflammatory injury, or nephrotic in nature, characterized by proteinuria.
![]() In the absence of specific and effective therapy for many types of glomerulonephritis, supportive treatments for edema, hypertension, hyperlipidemia, and intravascular thrombosis play important roles in reducing the complications associated with the disease.
In the absence of specific and effective therapy for many types of glomerulonephritis, supportive treatments for edema, hypertension, hyperlipidemia, and intravascular thrombosis play important roles in reducing the complications associated with the disease.
![]() To maximize therapeutic benefits and minimize drug-induced complications, patients have to be monitored closely to assess their therapeutic responses as well as the development of any treatment-induced toxicities.
To maximize therapeutic benefits and minimize drug-induced complications, patients have to be monitored closely to assess their therapeutic responses as well as the development of any treatment-induced toxicities.
![]() Among all the types of glomerulonephritis, minimal-change nephropathy is most responsive to treatment. Steroids can induce good responses in most patients during initial treatment as well as relapse.
Among all the types of glomerulonephritis, minimal-change nephropathy is most responsive to treatment. Steroids can induce good responses in most patients during initial treatment as well as relapse.
![]() Because of the lack of consistently effective treatment for primary focal segmental glomerular sclerosis, angiotensin-converting enzyme inhibitors or angiotensin receptor blockers are commonly used for patients with mild disease to control symptoms. Steroids and immunosuppressive agents are reserved for patients with severe disease.
Because of the lack of consistently effective treatment for primary focal segmental glomerular sclerosis, angiotensin-converting enzyme inhibitors or angiotensin receptor blockers are commonly used for patients with mild disease to control symptoms. Steroids and immunosuppressive agents are reserved for patients with severe disease.
![]() The optimal treatment for lupus nephritis depends on the underlying lesion and disease activity, as well as the severity and duration of the clinical presentation.
The optimal treatment for lupus nephritis depends on the underlying lesion and disease activity, as well as the severity and duration of the clinical presentation.
![]() The treatment of poststreptococcal glomerulonephritis is mainly supportive and symptomatic. Antibiotic therapy does not prevent subsequent diseases but may reduce the severity.
The treatment of poststreptococcal glomerulonephritis is mainly supportive and symptomatic. Antibiotic therapy does not prevent subsequent diseases but may reduce the severity.
The precise pathogenetic mechanisms of many glomerular diseases remain unknown, and the available therapeutic regimens are still far from optimal. This chapter provides an overview of the primary causes of glomerulonephritis with a focus on their etiology, the pathophysiologic mechanisms responsible for glomerular injury, and the clinical presentation of the eight predominant types of glomerulonephritis. Treatment options and monitoring approaches for each of these types of glomerulonephritis are also discussed. Diabetes mellitus is an important secondary cause of glomerular injury, and a thorough discussion of the pathophysiology and management of this condition can be found in Chapter 57.
NORMAL GLOMERULAR ANATOMY AND FUNCTION
The glomerulus, which is enclosed within the Bowman’s capsule, consists of two important components: the filtration barrier and the mesangium (Fig. 32-1). The capillary wall, which serves as a filtration barrier, consists of three well-defined layers: fenestrated endothelium, glomerular basement membrane (GBM), and epithelial cell layer. The epithelial cells, also known as podocytes, have specialized foot processes embedded in the outer layer of the GBM. It is across this barrier that fluid flows and ultimately becomes the ultrafiltrate. Under normal conditions, the GBM functions as a compact hydrated gel of matrix proteins with a pore-like structure. The mesangium, which consists of mesangial cells embedded in an extracellular matrix, provides support for the glomerular capillaries and also modulates blood flow through the capillaries.
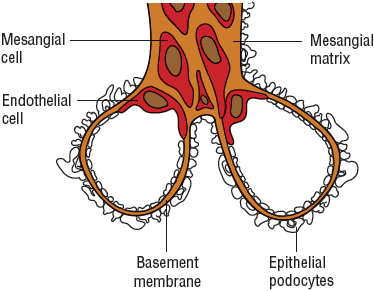
FIGURE 32-1 Microanatomy of the glomerulus.
The unique capillary bed of the glomerulus allows small nonprotein plasma constituents up to the size of inulin, which has a molecular weight of 5.2 kDa, to pass freely while excluding macromolecules equal to or larger than albumin, which has a molecular weight of 69 kDa. The ease of passage of solutes through the glomerular membrane is impacted by both the size and charge of the solute. Fixed, negatively charged sites are found within the glomeruli in all three layers of the capillary wall: the endothelium, the epithelium, and the GBM. The movement of negatively charged molecules is thus restricted more than that of neutral or positively charged molecules. Different glomerular diseases affect this size-and charge-selective barrier to different extents; consequently, glomerulopathies present with varied clinical features and solute-excretion patterns.
Some of the glomerular cells, such as the epithelial cells, have phagocytic function that can remove macromolecules trapped within the filtration barrier. They are also capable of synthesizing the GBM. In contrast, the mesangial cells regulate glomerular hemodynamics in response to angiotensin II and by producing prostaglandins. These cells also synthesize and respond to various cytokines and thus play a key role in immune-mediated glomerular diseases. Resident phagocytes in the mesangium are responsible for moving macromolecules trapped in the basement membrane into the urinary space. They are also involved in the development of both immune and nonimmune glomerular injury.
EPIDEMIOLOGY AND ETIOLOGY
In the United States in 2010, glomerulonephritis was the third most common cause of end-stage renal disease (ESRD), accounting for approximately 15% of all the living ESRD patients. About 7,300 patients (6.4% of all patients) develop stage 5 chronic kidney disease, which is also called ESRD, because of glomerulonephritis each year.1
Humoral and cellular immunologic mechanisms participate in the pathogenesis of most glomerulonephritis. Abnormalities in coagulation and metabolism, as well as hereditary and vascular diseases, also contribute to glomerular damage. The histopathologic manifestations vary substantially among the different types of glomerulonephritis. An overview of the primary pathogenetic mechanisms is presented in this section, and specific abnormalities for each of the primary types of glomerulonephritis are presented in subsequent sections.
PATHOPHYSIOLOGY
![]() The glomerular lesion may be diffuse (involving all glomeruli), focal (involving some but not all glomeruli), or segmental, also known as local (involving part of the individual glomerulus). The pathologic manifestations may also be described as proliferative (overgrowth of epithelium, endothelium, or mesangium), membranous (thickening of GBM), and/or sclerotic.
The glomerular lesion may be diffuse (involving all glomeruli), focal (involving some but not all glomeruli), or segmental, also known as local (involving part of the individual glomerulus). The pathologic manifestations may also be described as proliferative (overgrowth of epithelium, endothelium, or mesangium), membranous (thickening of GBM), and/or sclerotic.
The glomerular capillary wall is particularly susceptible to immune-mediated injury. Antigens and antibodies tend to localize in the glomerulus, probably because of its high blood flow and capillary hydrostatic pressure. Parenchymal damage can be induced as a result of humoral- and cell-mediated immune reactions. Antibodies and sensitized T lymphocytes are the primary mediators of glomerular injury.2,3
Production of antibodies to endogenous or exogenous antigens that are recognized as foreign by the host is the first step in humoral immunologic damage to the glomerulus. Endogenous antigens may be intrinsic glomerular antigens, such as Heymann antigen on the epithelial cell or Goodpasture antigen on the GBM, or previously sequestered antigens, such as DNA or thyroglobulin. Exogenous antigens are most often viral, bacterial, parasitic, or fungal in origin. Antineutrophil cytoplasmic autoantibodies (ANCAs) (i.e., autoantibodies that react to the cytoplasmic components of neutrophils and monocytes) are found in patients with idiopathic crescentic glomerulonephritis and also in the accompanying vasculitis.
Complexes of antigens and antibodies may be formed in the circulation and then passively entrapped in the glomerular capillary or mesangium. Alternately, experimental antibodies may combine with endogenous glomerular antigens or exogenous antigens entrapped in the glomerulus to form complexes locally, or in situ.3 The type and extent of glomerular damage depend on the location of the immune complex formation and the rate at which it is removed. Impaired removal facilitates the growth of the complex and thus increases the likelihood of glomerular damage.
Subsequent to antigen–antibody formation, a series of biologic events is triggered that ultimately leads to glomerular injury. Noninflammatory lesions can result from the binding of noncomplement-fixing antibody to the glomerular epithelial cell (mechanism 1) or from the activation of the complement system to form the C5b-9 membrane attack complex (mechanism 2).3 Both mechanisms can damage the glomerular epithelial cell and result in capillary wall injury and proteinuria. Inflammatory lesions are induced by glomerular infiltration of circulating inflammatory cells such as neutrophils, monocytes/macrophages, and platelets (mechanism 3) or by proliferation of resident glomerular mesangial cells (mechanism 4), resulting in GBM damage.3 The migration of neutrophils and monocytes to the glomerular tufts is promoted by chemoattractants such as complement fragments (C3a and C5a), platelet-activating factor, interleukin-8, and monocyte chemotactic protein-1.4 Various cytokines, chemokines, and growth factors are then released to participate in the inflammatory process.2
T cells sensitized to glomerular antigen, macrophages, and resident mesangial cells are important participants in cell-mediated injury. Sensitized T cells can cause glomerular hypercellularity in the absence of antibody deposition.2–4 Cytotoxic T cells may bind with the target cells and destroy them. Alternatively, a delayed-type hypersensitivity reaction may be initiated by activated T cells through the release of lymphokines to attract, activate, and transform monocytes into macrophages.3 These humoral and cellular mediators, in conjunction with a host of toxic molecular entities including reactive oxygen species, proteinases, eicosanoids, and procoagulants, which are secreted by neutrophils, macrophages, platelets, and resident glomerular cells, can alter the permeability, blood flow, and function of the glomeruli. Vascular constriction and occlusion follow and result in the eventual destruction of the glomeruli.
Acute forms of glomerular injury frequently lead to chronic and persistent renal dysfunction, even though the original immune factors that induced the initial glomerular injury have resolved. Experimental and clinical investigations suggest that a variety of factors may participate in the progression of renal injury. These factors include systemic and glomerular hypertension, high dietary protein intake, proteinuria, glomerular hypertrophy, hyperlipidemia, activation of the coagulation system, abnormalities of calcium and phosphorus balance, and tubulointerstitial injury. The degree of proteinuria not only is an index of the severity of glomerular disease but also has been associated with an increased rate of progression of renal injury. Heavy proteinuria is an indicator of poor prognosis in various glomerular diseases.
Proteinuria is also accompanied by an increased flux of macromolecules across the mesangium. The mesangial overload may then lead to structural damage. The passage of serum components, such as complement, across the GBM may have a pathophysiologic effect on the glomerular epithelial cells and alter the integrity of the glomerular filtration barrier. The damaging effects of macromolecules other than albumin, such as immunoglobulins, lipoproteins, transferrin, and complement, remain to be characterized.
CLINICAL PRESENTATION
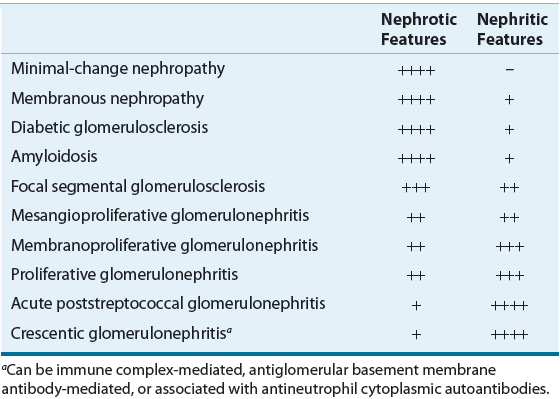
![]() Although patients with glomerular disease may present with an array of signs and symptoms, they are often categorized into one of two broad classifications: nephritic syndrome or nephrotic syndrome (Table 32-1). The unique clinical presentation characteristics of the predominant glomerulopathies are described in the individual disease sections, presented later in the chapter.
Although patients with glomerular disease may present with an array of signs and symptoms, they are often categorized into one of two broad classifications: nephritic syndrome or nephrotic syndrome (Table 32-1). The unique clinical presentation characteristics of the predominant glomerulopathies are described in the individual disease sections, presented later in the chapter.
TABLE 32-1 Tendencies of Glomerular Diseases to Manifest Nephrotic and Nephritic Features
Nephritic syndrome reflects glomerular inflammation and frequently results in hematuria. White cells and cellular and granular casts are commonly found in the urine. In contrast, nephrotic syndrome reflects noninflammatory injury to the glomerular structures and results in few cells or cellular casts in the urine. Initially, there may be limited or no reduction in renal excretory function.
CLINICAL PRESENTATION Nephritic and Nephrotic Syndromes
Hematuria occurs when red blood cells leak through the openings of the GBM. The presence of red cell casts is highly indicative of glomerulonephritis or vasculitis. The presence of dysmorphic red blood cells in the urine is suggestive of glomerular disease. The red blood cells are damaged as they pass through the openings in the GBM or the cells may sustain osmotic injury as they travel through the different osmotic environments within the lumen of the kidney tubules.
The presence of proteinuria indicates a defect of the size-and/or charge-selective barriers within the GBM. Normal urinary protein excretion is between 40 and 80 mg/day, with a maximum of 150 mg. Fewer than 20 mg of the excreted proteins are albumin. Most of the albumin that enters the glomerular filtrate is either reabsorbed or catabolized by the tubular epithelium. The dipsticks that are commonly used to identify proteinuria detect only albumin; they become positive when protein excretion is more than 300 to 500 mg/day. They are therefore unable to detect the early stages of renal injury secondary to diabetes mellitus or hypertension, which often result in microalbuminuria with urinary albumin excretion ranges between 30 and 300 mg/day. Chemstrip Micral-Test II (Roche Diagnostics, Indianapolis, IN), a simple immunoassay on a dipstick, permits specific and semiquantitative determination of urinary albumin concentrations at five levels: 0, 10, 20, 50, and 100 mg/L. Another qualitative test, Micro-Bumintest (Bayer Diabetes Care, Mishawaka, IN), registers a positive reading when the urine albumin concentration is greater than 40 mg/L.
Hypertension is common for patients with glomerular diseases, as a result of renal salt retention causing plasma volume expansion. In contrast, increased activity of vasoconstrictors such as angiotensin II is often the cause for patients with chronic glomerular diseases. Scarring of the glomerulus resulting in regional ischemia is thought to be responsible for the hypertension. Activation of the sympathetic nervous system and the release of vasoconstrictor substances may also contribute.
Nephritic Syndrome
Glomerular bleeding resulting in hematuria is typical in nephritic syndrome. Dysmorphic red cells, especially acanthocytes, are a sensitive and specific marker of glomerular bleeding. The presence of pus and cellular and granular casts in the urine is common. The extent of proteinuria is variable. Patients with severe nephritic glomerular injury have renal function impairment because of the reduced glomerular surface area available for filtration, as a result of constriction of the capillary lumen by proliferating mesangial cells or inflammatory cells.
Nephrotic Syndrome
Nephrotic syndrome is characterized by proteinuria greater than 3.5 g/day per 1.73 m2, hypoproteinemia, edema, and hyperlipidemia. A hypercoagulable state may also be present in some patients. The syndrome may be the result of primary diseases of the glomerulus, or be associated with systemic diseases such as diabetes mellitus, lupus, amyloidosis, and preeclampsia. Hypoproteinemia, especially hypoalbuminemia, results from increased urinary loss of albumin and an increased rate of catabolism of filtered albumin by proximal tubular cells. The compensatory increase in hepatic synthesis of albumin is insufficient to replenish the protein loss, probably because of malnutrition.
Edema formation in patients with nephrotic syndrome was traditionally thought to be driven by the reduced plasma oncotic pressure secondary to hypoalbuminemia. If the oncotic pressure is low, the movement of fluid from the vascular space to the interstitial compartment results in a reduction of the plasma volume, which can trigger compensatory renal sodium and water retention through the activation of the renin–angiotensin–aldosterone axis, vasopressin, and the sympathetic nervous system (the “underfill” mechanism). However, experimental data reveal that the plasma volume is actually normal or elevated. Hypoalbuminemia may not cause edema until the serum albumin concentration is less than 2 g/dL (20 g/L). In addition, the transcapillary oncotic pressure gradient is not as high as previously thought because increased lymphatic flow reduces the interstitial oncotic pressure by removing protein and fluid from the interstitium, thereby reducing the transcapillary oncotic pressure gradient.5 Instead, fluid retention is likely mediated by a primary increase in sodium reabsorption at the distal nephron, which is probably caused by tubular resistance to the action of atrial natriuretic peptide (the “overflow” mechanism).6 It is likely that both mechanisms may contribute to nephrotic edema in different patients.6
Albuminuria greater than 3 g daily is associated with a significant increase in serum cholesterol concentrations for patients with primary glomerular disease.7 Hyperlipidemia in nephrotic syndrome is characterized by elevated serum total cholesterol and triglyceride concentrations, with increased very-low-density lipoprotein (VLDL) and low-density lipoprotein (LDL) cholesterol concentrations. Lipoprotein (a) levels may also be increased. The reduced plasma oncotic pressure as a result of hypoalbuminemia may stimulate hepatic synthesis of lipids and lipoproteins. The increased VLDL production and increased liver cholesterol synthesis, along with a decrease in LDL receptor activity, can then lead to an increase in LDL cholesterol concentrations. In addition, reduced serum albumin or the loss of a liporegulatory substance may result in reduced VLDL clearance.8 Nephrotic patients with hyperlipidemia, especially those with concomitant hypertension, are presumed to have an increased risk for atherosclerotic vascular disease. Hyperlipidemia also promotes the progression of glomerular injury, as evidenced by glomerulosclerosis, mesangial expansion, and hyalinosis.8,9
Many patients with nephrotic syndrome have a hypercoagulable state caused by defects of several control proteins in the coagulation cascade. The concentration of the coagulation inhibitor antithrombin III is reduced because of increased loss in the urine. A reduced amount of the coagulation inhibitors proteins C and S, along with increased concentrations of factors V and VIII, increased fibrinogen concentrations, and abnormal platelet function, may also contribute to the hypercoagulable state. The net result of these alterations in coagulation is an increased risk for arterial and venous thrombosis, especially in the deep veins and renal veins. As many as 25% of patients with membranous nephropathy may have renal vein thrombosis.
DIAGNOSTIC CONSIDERATIONS
Patients with suspected glomerular disease should have an extensive medical history obtained to identify potential systemic causes (Table 32-2). Medication, environmental, and occupational histories may also help identify possible exposure to potentially nephrotoxic agents. A carefully conducted physical examination and laboratory evaluation may reveal the presence of systemic diseases that may contribute to the development of glomerular disease (Fig. 32-2). In addition, the patient’s age, gender, and ethnic background may be helpful in pinpointing the specific type of glomerular disease. Many of the conditions are more prevalent in certain age groups, although they may occur at any age. For example, proliferative glomerulonephritis is more common in those younger than 40 years of age, whereas the incidence of membranous glomerulonephritis is dramatically higher in those older than 50 years of age.
TABLE 32-2 Evaluation of Patients Suspected of Having Glomerular Disease

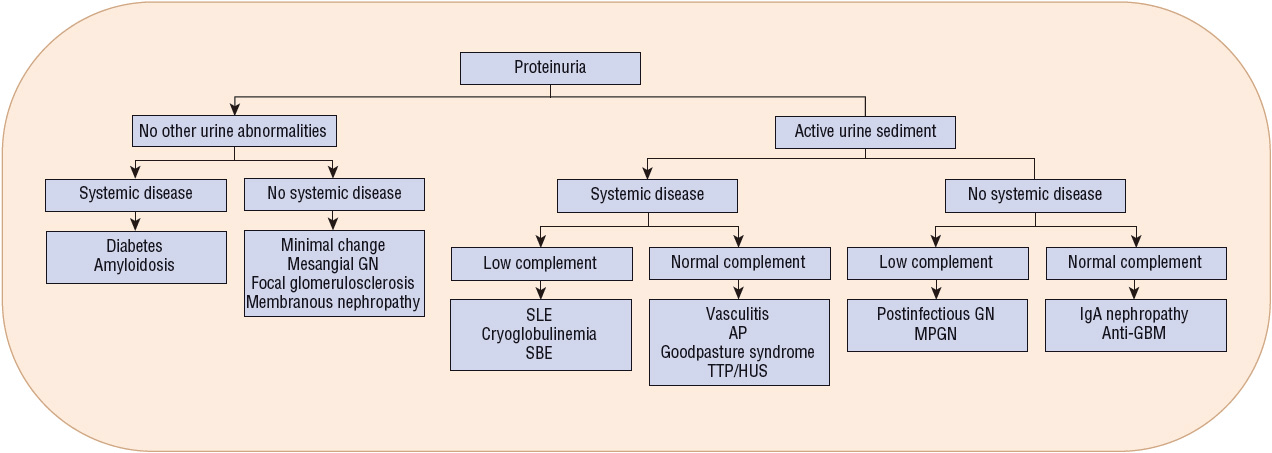
FIGURE 32-2 Clinical presentations of glomerulonephritis. (AP, anaphylactoid purpura; GBM, glomerular basement membrane; GN, glomerulonephritis; HUS, hemolytic uremic syndrome; IgA, immunoglobulin A; MPGN, membranoproliferative glomerulonephritis; SBE, subacute bacterial endocarditis; SLE, systemic lupus erythematosus; TTP, thrombotic thrombocytopenic purpura.)
Laboratory evaluation such as urinalysis can help differentiate the nephrotic or nephritic nature of the disease. The glomerular filtration rate (GFR) may be used to determine the extent of glomerular damage. In the early stages of the disease, the GFR may remain normal. Initial injury to the glomerulus primarily lowers the permeability coefficient (Kf) of the GBM by reducing the surface area available for filtration and/or the unit permeability of the membrane. The reduced permeability is compensated by an elevation in the glomerular capillary hydrostatic pressure through afferent arteriolar dilation and efferent arteriolar constriction. Extensive glomerular damage may therefore be present before a substantial reduction of total GFR is evident.
Although the cause of glomerular disease may be established from clinical and laboratory evaluation, sometimes percutaneous renal biopsy may be needed to provide a definitive diagnosis.
TREATMENT
General Approach to Treatment
The course and prognosis of the different glomerular diseases are extremely variable and depend on the underlying etiology. In glomerular diseases with a secondary cause, such as poststreptococcal glomerulonephritis (PSGN), after the initiating factor is removed, the prognosis of the renal disease is often good. In contrast, the rates of renal function deterioration among the primary glomerulonephritides vary markedly. The majority of patients with minimal-change disease, IgA nephropathy, and membranous nephropathy have a fairly good prognosis. However, those with focal segmental glomerulosclerosis (FSGS) who are resistant to therapy, as well as those with rapidly progressive glomerulonephritis (RPGN) who are untreated, are likely to experience rapid loss of renal function. In some instances, half of the renal function may be lost within a 3-month period. Certain glomerulonephritides, such as minimal-change nephropathy, are very responsive to treatment while patients with membranous proliferative glomerulonephritis are rarely responsive to existing therapies.
Because of the variable courses exhibited by the different glomerulonephritides, specific treatment approaches have been developed for each disease. When natural history of the glomerulonephritis is well delineated, it is more likely that potential regimens can be designed and evaluated from both therapeutic and economic perspectives. The potential therapeutic benefits of treatment regimens should always be weighed against the risks to which the patients are being exposed. It is therefore imperative to identify patients who are most likely to benefit from treatment, especially those who have other risk factors that may contribute to the deterioration of their renal function. In those instances in which satisfactory regimens are not available to treat the primary disease, appropriate supportive measures should be employed. Optimization of systemic and glomerular blood pressure, reducing proteinuria, and possibly controlling hyperlipidemia may all improve the long-term outcome as well as the quality of life of these patients.
KDIGO (Kidney Disease: Improving Global Outcomes) is a global nonprofit foundation dedicated to improving the care and outcomes of kidney disease patients worldwide through promoting coordination, collaboration, and integration of initiatives to develop and implement clinical practice guidelines for many kidney diseases through its work groups of experts.10 The quality of evidence and strength of recommendations were graded. Many of these clinical practice guidelines are referenced in the ensuing sections on the treatment of individual primary glomerular diseases.
Nonpharmacologic Therapy
![]() For patients with nephrotic syndrome, dietary measures involve restriction of sodium intake to 50 to 100 mEq/day (50 to 100 mmol/day),11 protein intake of 0.8 to 1 g/day,11,12 and a low-lipid diet of less than 200 mg cholesterol. Total fat should account for less than 30% of daily total calories.11 Sodium restriction is important not only in the control of edema, but also in the control of hypertension and proteinuria. Similarly, protein restriction not only helps to reduce proteinuria but also has a potential role in retarding the progression of renal disease. Patients should also stop smoking because a dose-dependent increase in risk for developing ESRD was observed in men with primary inflammatory (immunoglobulin A glomerulonephritis) or noninflammatory (polycystic kidney disease) renal diseases.13
For patients with nephrotic syndrome, dietary measures involve restriction of sodium intake to 50 to 100 mEq/day (50 to 100 mmol/day),11 protein intake of 0.8 to 1 g/day,11,12 and a low-lipid diet of less than 200 mg cholesterol. Total fat should account for less than 30% of daily total calories.11 Sodium restriction is important not only in the control of edema, but also in the control of hypertension and proteinuria. Similarly, protein restriction not only helps to reduce proteinuria but also has a potential role in retarding the progression of renal disease. Patients should also stop smoking because a dose-dependent increase in risk for developing ESRD was observed in men with primary inflammatory (immunoglobulin A glomerulonephritis) or noninflammatory (polycystic kidney disease) renal diseases.13
Because many immune factors are implicated in the pathogenesis of glomerulonephritis, plasmapheresis may be used to remove these mediators. During the procedure, whole blood is removed from the body and centrifugation is used to separate the cellular elements from the plasma. The cells are then infused back to the patient after resuspension in saline or plasma substitute. The plasma proteins, presumably including the pathogenic immune factors, are thereby removed from the patient.
Pharmacologic Therapy
Immunosuppressive Agents
Immunosuppressive agents, alone or in combination, are commonly used to alter the immune processes that are responsible for the glomerulonephritides. Corticosteroids, in addition to their immunosuppressive effect, also possess antiinflammatory activities. They reduce the production and/or release of many substances that mediate the inflammatory process, such as prostaglandins, leukotrienes, platelet-activating factors, tumor necrosis factors, and interleukin-1 (IL-1). Movement of leukocytes and macrophages to the site of inflammation is also inhibited. The immunosuppressive effects of corticosteroids are mediated through the inhibition of the release of IL-1 and tumor necrosis factor by activated macrophages, and interleukin-2 by activated T cells. In addition, the actions of migration-inhibiting factor and γ-interferon are inhibited. Processing of antigens is thus affected by the presence of corticosteroids. Cytotoxic agents, such as cyclophosphamide, chlorambucil, or azathioprine, are commonly used to treat glomerular diseases. Cyclosporine can reduce lymphokine production by activated T lymphocytes, and it may decrease proteinuria by improving the permselectivity of the GBM. Mycophenolate mofetil is useful in different glomerulonephritides because of its effects on T- and B-cell lymphocytes.
Recently, many novel targets were identified and new agents are being evaluated for their usefulness to control the disease, preserve renal function, and improve patient outcome.14 To stay abreast of the expanding availability of treatment options, one can routinely consult one of the clinical trial registries, such as www.clinicaltrials.gov.
Diuretics
Management of nephrotic edema involves salt restriction, bed rest, and use of support stockings and diuretics. However, severe salt restriction is difficult to achieve and prolonged bed rest can predispose nephrotic patients to thromboembolism. Hence the use of a loop diuretic such as furosemide is frequently required. Although the delivery of diuretic to the kidney tubules is normal, the presence of large amounts of protein in the urine promotes drug binding, and thereby reduces the availability of the diuretic to the luminal receptor sites. In addition, reduced sodium delivery to the distal tubule secondary to decreased glomerular perfusion may also alter diuretic effectiveness. Large doses of the loop diuretic, such as 160 to 480 mg of furosemide, may be needed for patients with moderate edema (see Chap. 34). In some instances, a thiazide diuretic or metolazone may be added to enhance natriuresis.11,15 Alternatively, continuous IV infusion of a loop diuretic, such as furosemide 160 to 480 mg/day, may be employed.16 For patients with morbid edema, albumin infusion may be used to expand plasma volume and increase diuretic delivery to the renal tubules, thus enhancing diuretic effect. However, it may precipitate congestive heart failure and may also reduce therapeutic response to steroid in minimal-change nephropathy. For patients with significant edema, the goal of treatment should be a daily loss of 1 to 2 lb (0.45 to 0.9 kg) of fluid until the patient’s desired weight has been obtained.
Antihypertensive Agents
Optimal control of hypertension for patients with glomerular disease is important in reducing both the progression of renal disease and the risk for cardiovascular disease12 (see Chaps. 3 and 29). The target blood pressure for patients with chronic kidney disease defined by GFR <60 mL/min (<1 mL/s) or albuminuria >300 mg/day is less than 130/80 mm Hg.17 Angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin II receptor blockers (ARBs) delay the loss of renal function for patients with diabetic and nondiabetic (primarily glomerulonephritis) renal diseases.18 Nondihydropyridine calcium channel blockers (e.g., diltiazem, verapamil) reduce proteinuria and preserve renal function and could be used as an additional agent. In contrast, the dihydropyridine calcium channel blockers (e.g., nifedipine, amlodipine, or nisoldipine) are effective in lowering blood pressure, but without the benefit of proteinuria reduction.19
Antiproteinuria Agents
Dietary protein restriction reduces proteinuria and may retard renal function deterioration. Secondary analysis of the Modification of Diet in Renal Disease Study for patients with moderate renal insufficiency (GFR of 25 to 55 mL/min/1.73 m2 [0.24 to 0.53 mL/s/m2]) revealed that reduced protein intake (0.66 g/kg/day) delayed the rate of GFR deterioration for patients with severe renal insufficiency (GFR of 13 to 24 mL/min/1.73 m2 [0.13 to 0.23 mL/s/m2]).20 Consequently, modest protein restriction of 0.8 g/kg/day is reasonable for patients with moderate renal insufficiency. Decreasing dietary protein also reduces the intake of phosphorus and potassium. In many instances, the potential benefits of protein restriction have to be balanced against the need for protein intake to overcome nutritional deficiencies. For nondialyzed patients who have GFRs of less than 25 mL/min/1.73 m2 (0.24 mL/s/m2), dietary protein intake should be reduced to 0.6 g/kg/day.13
Angiotensin-Converting Enzyme Inhibitors and Receptor Blockers It is now recognized that proteinuria is an independent risk factor for renal function decline and cardiovascular disease.21 Reducing proteinuria can retard renal function loss and delay the progression to ESRD.22 The antiproteinuric effect of ACEIs is associated with a fall in filtration fraction, suggesting a reduction in intraglomerular pressure. Recent studies show that ACEIs and ARBs may also have direct effects on podocytes, resulting in reduction of proteinuria and glomerular scarring.23 In addition, angiotensin-converting enzyme (ACE) inhibition may also reduce the effect of angiotensin II on renal cell proliferation, thereby reducing sclerosis. These beneficial effects on proteinuria are beyond what can be attributed by the drug’s antihypertensive effects (see Chaps. 3 and 29).24,25
Clinical Controversy…
The combined use of an ACEI and an ARB reduces the rate of renal function decline more than either treatment alone.18 A meta-analysis of 21 randomized, controlled studies revealed that combination therapy enhanced the reduction of proteinuria in both diabetic and nondiabetic patients.26 Combination therapy maximizes blockade of the renin–angiotensin system by counteracting the effects of angiotensin II produced by non-ACE pathways. In addition, with the blockade of the angiotensin II type 1 receptor, the angiotensin II produced by the non-ACE pathways may still act on the angiotensin II type 2 receptors, further facilitating vasodilation.27 An angiotensin II receptor antagonist should therefore be added to the regimen for those patients who do not attain full and persistent remission of proteinuria with an ACEI alone. A thorough review of the combined use of ACEs and ARBs for diabetic nephropathy and proteinuria reduction is found in Chapter 29.
Nonsteroidal Antiinflammatory Agents Nonsteroidal antiinflammatory drugs (NSAIDs) probably reduce proteinuria through prostaglandin E2 inhibition, resulting in a reduction of intraglomerular pressure, a decrease in GFR, and restoration of the barrier size selectivity of the GBM.12 Indomethacin and meclofenamate are the two most evaluated NSAIDs. Their antiproteinuric effect is comparable to that attained with ACEIs, and combined treatment with an ACEI results in additional proteinuria reduction.28 However, adherence to a low-sodium diet or concurrent use of a diuretic is needed to maximize the antiproteinuric effect. Because of their potential for nephrotoxicity, especially for patients with poor renal function, long-term use of an NSAID for renoprotection is not preferred.24
Adrenocorticotropin A synthetic adrenocorticotropin (ACTH) analog has been used in Europe for proteinuria reduction associated with nephrotic syndrome. It was reported to have effects similar to alternating months of steroids and cyclophosphamide.29 Instead of the synthetic analog, a natural, purified ACTH gel is available in the United States and is approved by the FDA for inducing a remission of proteinuria in the nephrotic syndrome without uremia of the idiopathic type or that due to lupus erythematosus. Favorable response was reported in an observation series of 21 patients in the United States.30 However, the authors cautioned the interpretation of the results since the data were not derived from a controlled, randomized study. The patients had different glomerular diseases and the long-term effect was not reported.
Statins
An abnormal lipoprotein profile increases the risk of atherosclerosis and coronary heart disease for patients with nephrotic syndrome. It is therefore important to treat patients with persistent nephrotic syndrome and sustained dyslipidemia, especially those with high VLDL and LDL cholesterol levels in the presence of a normal or low high-density lipoprotein cholesterol level (see Chaps. 11 and 29). Therapy is especially needed for those with concurrent atherosclerotic cardiovascular disease, or with additional risk factors for atherosclerosis, such as smoking and hypertension.8
A low-fat diet is usually not sufficient to correct hyperlipoproteinemia.12 β-Hydroxy-β-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors, also known as “statins” such as lovastatin, pravastatin, simvastatin, and fluvastatin, are considered the treatment of choice.12 They reduce total plasma cholesterol concentration, LDL cholesterol, and total plasma triglyceride concentrations.8 Aside from the lipid-lowering effects, statins may confer renoprotection through different mechanisms, including reduction of cell proliferation and mesangial matrix accumulation and antiinflammatory and immunomodulatory effects.31 Recent clinical studies show that they can reduce proteinuria and delay renal function loss.32,33 The combined use of an ACEI with a statin may offer additional benefits in controlling nephrotic hyperlipidemia.20
Anticoagulants
Renal vein thrombosis, pulmonary emboli, or other thromboembolic events are serious and common complications of nephrotic syndrome, and are frequently seen in those with membranous nephropathy. Although patients who have documented thromboembolic episodes should be anticoagulated with warfarin until remission of nephrotic syndrome, the use of prophylactic anticoagulation is controversial. A decision analysis study suggested that prophylactic anticoagulation is beneficial for patients with membranous nephropathy.34 Prophylactic anticoagulation is not recommended for all patients; rather, a “selective” approach or individualized assessment should be conducted to identify those at high risk (i.e., those with severe nephrotic syndrome and a serum albumin concentration <2 to 2.5 g/dL [<20 to 25 g/L]).34 Also at risk are those who require prolonged bed rest, those receiving high-dose IV steroid therapy, and individuals who are dehydrated as well as postsurgical patients.12
Evaluation of Therapeutic Outcomes
The management of patients with glomerulonephritis involves specific pharmacologic therapy for the glomerular disease and supportive measures to prevent and/or treat the pathophysiologic sequelae, namely, hypertension, edema, and progression of renal disease. Although the course of the disease, as well as the specific treatment regimens, varies among the different glomerulonephritis, the monitoring parameters for efficacy assessment are similar. For patients with nephrotic syndrome, supportive therapy should also address the management of extrarenal complications of heavy proteinuria, namely, hypoalbuminemia, hyperlipidemia, and thromboembolism. Because patients with significant proteinuria tend to have more rapid decline of renal function, reduction of proteinuria thus becomes critical in delaying the rate of progression toward ESRD.
![]() Patients should be monitored closely for therapeutic response as well as the development of treatment-related toxicities. Although the rate of renal function deterioration is an important indicator of the long-term success of treatment, resolution of nephrotic and nephritic signs and symptoms associated with the glomerulopathies is an important short-term therapeutic target (see Table 32-3).
Patients should be monitored closely for therapeutic response as well as the development of treatment-related toxicities. Although the rate of renal function deterioration is an important indicator of the long-term success of treatment, resolution of nephrotic and nephritic signs and symptoms associated with the glomerulopathies is an important short-term therapeutic target (see Table 32-3).
TABLE 32-3 Monitoring Parameters to Assess Response to Glomerulonephritis Treatment

Serum creatinine concentration as well as creatinine clearance should be evaluated prior to and during treatment; 24-hour urine outflow should be collected to determine the extent of proteinuria. Alternatively, the daily urine protein excretion may be estimated from the urinary total protein-to-creatinine concentration ratio. After establishing the correlation between the 24-hour urinary protein excretion and the protein-to-creatinine ratio, single, random urine specimens may be used in place of a 24-hour urine collection. Blood pressure should be monitored periodically to assess the need for and the adequacy of antihypertensive therapy. The pressures should also be evaluated in conjunction with clinical signs and symptoms of edema and fluid overload to gauge the need for volume control as well as diuretic use. For patients with nephrotic syndrome, serum lipid concentrations should be monitored. If the patient has hematuria, urinalysis and a complete blood count should be obtained. The clinician should also be aware of the patient’s appetite and energy level, because these are indicators of the patient’s overall state of well-being. At times, renal biopsy is needed to assess response to treatment and disease progression, to determine future treatment strategy, and to confirm the initial diagnosis.
Patients receiving cytotoxic drug treatment should be evaluated for drug-related toxicities every week during the initial treatment period. After 1 month of treatment, the frequency of monitoring may be reduced. When the patient is on long-term steroid treatment, monthly visits are often required for assessment of both efficacy and toxicities. If a favorable response is obtained after a course of treatment, the patient may be evaluated every 3 to 4 months. The patient’s renal function, proteinuria, urinalysis, blood pressure, lipid profile, and the overall state of health should be assessed during these regular follow-up visits.
Minimal-Change Nephropathy
Epidemiology and Etiology
Minimal-change nephropathy (also termed nil disease or minimal-change disease) is commonly found in children, accounting for about 85% to 90% of all cases of nephrotic syndrome in children between 1 and 4 years of age. The percentage drops gradually to less than 50% after age 10 years and accounts for less than 20% of all cases of idiopathic nephrotic syndrome in adults. Secondary causes of minimal-change nephropathy include drug administration (e.g., NSAIDs, lithium, interferons), lupus, and various T-cell–related disorders, such as Hodgkin’s disease and leukemias.
Pathophysiology
Minimal-change disease is characterized by the absence of definitive pathologic changes observed under light and immunofluorescence microscopy. The characteristic lesion in patients with minimal-change disease, as visualized under electron microscopy, is the spreading and fusion of the foot processes of epithelial cells over an unchanged GBM. Lipoid nephrosis is another term that has been used to describe this type of glomerular disease because lipids, as well as renal tubular cells, are found in the urine. The pathogenesis of minimal-change disease is unknown. Altered cell-mediated immunologic response, specifically T-cell dysfunction or changes in the T-cell subpopulations, may be responsible. The activated lymphocytes are thought to secrete lymphokines that reduce the production of anions in the GBM. The permeability of the GBM to plasma albumin is increased through a reduction of electrostatic repulsion. The loss of anionic charges also results in fusion of the epithelial cell foot processes. Other vascular permeability factors, such as hemopexin, interleukin-4, and vascular endothelial growth factor, also have been suggested to be responsible.
Clinical Presentation
Most patients present initially with edema, frequently acute in onset, following a nonspecific upper respiratory tract infection, allergic reaction, or vaccinations, which might have activated the T lymphocytes. Nephrotic syndrome with massive proteinuria (substantially more than 40 mg/m2/h for children and >3 to 3.5 g/day for adults), edema, hypoalbuminemia, and hyperlipidemia is common. The patient’s weight may increase dramatically because of sodium and fluid retention. Nephritic features, such as gross hematuria, are uncommon. Hypertension and decreased renal function are uncommon in children but are more common in older adults.35 For some patients, volume depletion may result in mild-to-moderate azotemia.
TREATMENT
Pharmacologic Therapy
Steroids
![]() Minimal-change disease is most responsive to initial treatment with corticosteroids. In children, steroid therapy is expected to reduce proteinuria in approximately 90% of the patients, with >95% 10-year renal survival. Because of the excellent response to initial therapy with steroids and the prevalence of this glomerular disease in children, reduction of proteinuria secondary to steroid treatment is considered diagnostic for minimal-change disease without the need for biopsy. Prednisone is commonly administered at 60 mg/m2 per day initially for 4 to 6 weeks. The dose is then reduced to 40 mg/m2 per day every other day for another 4 to 6 weeks, with or without tapering afterward (Fig. 32-3).36 Proteinuria will disappear in 50% of patients after 1 week and in 90% of patients after 4 weeks of treatment. Different versions of the steroid regimen are available as there is no consensus on the optimal dose and duration. Studies are being conducted to identify the best strategy to induce remission, reduce disease recurrence, and minimize adverse effects of the therapy. Commonly, the initial episode is treated with an extended course (months) of therapy, followed by shorter treatment (weeks) for relapses.37
Minimal-change disease is most responsive to initial treatment with corticosteroids. In children, steroid therapy is expected to reduce proteinuria in approximately 90% of the patients, with >95% 10-year renal survival. Because of the excellent response to initial therapy with steroids and the prevalence of this glomerular disease in children, reduction of proteinuria secondary to steroid treatment is considered diagnostic for minimal-change disease without the need for biopsy. Prednisone is commonly administered at 60 mg/m2 per day initially for 4 to 6 weeks. The dose is then reduced to 40 mg/m2 per day every other day for another 4 to 6 weeks, with or without tapering afterward (Fig. 32-3).36 Proteinuria will disappear in 50% of patients after 1 week and in 90% of patients after 4 weeks of treatment. Different versions of the steroid regimen are available as there is no consensus on the optimal dose and duration. Studies are being conducted to identify the best strategy to induce remission, reduce disease recurrence, and minimize adverse effects of the therapy. Commonly, the initial episode is treated with an extended course (months) of therapy, followed by shorter treatment (weeks) for relapses.37
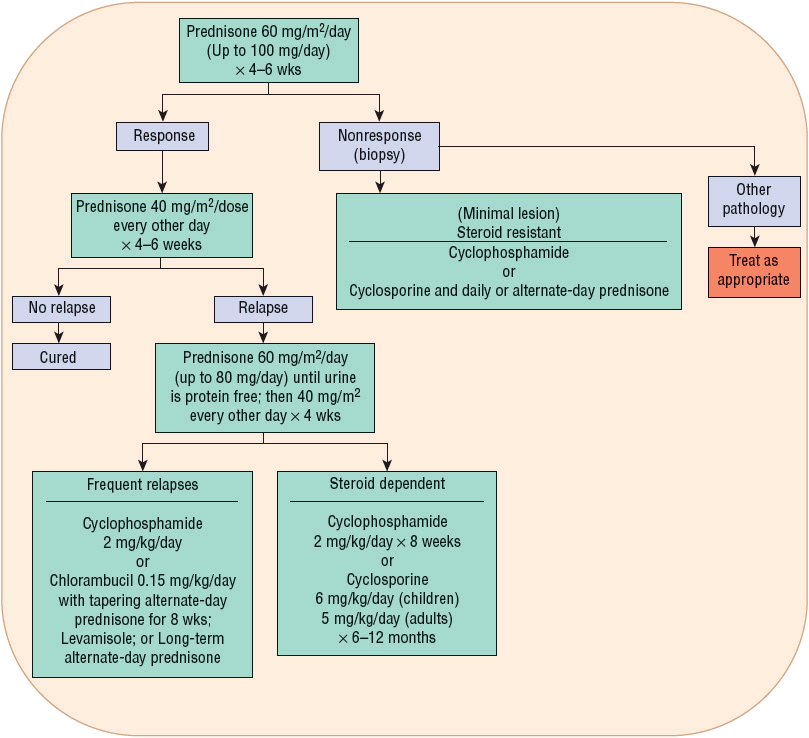
FIGURE 32-3 Treatment algorithm for minimal-change nephropathy. (Reprinted by permission from Macmillan Publishers Ltd: from reference 36.)
For adults, prednisone 1 mg/kg per day is given initially for 4 weeks with a reduction to 0.75 mg/kg every other day for the next 4 weeks. Proteinuria will disappear in 50% to 60% of patients after 8 weeks of treatment, and complete remission will be attained in 80% of patients after 28 weeks of therapy.35
Relapse As many as 85% of the patients who respond to initial steroid therapy (steroid sensitive) will experience a relapse of proteinuria, mostly within 6 to 12 months after disease onset. The risk of relapse is affected by the duration of initial steroid therapy.12,36 Children who were asymptomatic with proteinuria diagnosed during routine urine screening tend to have less frequent relapses and a more favorable clinical course. In those who relapse, 50% to 65% may have steroid-responsive relapse episodes over the subsequent 3- to 5-year period. The dose and duration of steroid treatment for the relapse do not influence the subsequent rate of relapse.12,36 Commonly, 60 mg/m2 per day of prednisone is given until the urine is free of protein for 3 days, to be followed by 4 weeks of alternate-day prednisone at 40 mg/m2 per dose.36
Frequent Relapse Approximately 10% to 20% of children that are responsive to steroid will experience three or four relapses. Half of them will then relapse frequently and become steroid dependent, requiring continuous low-dose alternate-day prednisone to maintain an extended relapse-free period.36 A small number of patients eventually develop resistance to steroids, and a biopsy done at that time often reveals another pathology such as FSGS. It is controversial whether minimal-change disease progresses into FSGS or whether the glomerulosclerosis that was present at the time of initial diagnosis was inadvertently diagnosed as minimal-change nephropathy because of tissue-sampling error during the renal biopsy.
Cytotoxic Agents
Cytotoxic agents are often considered for patients who are steroid resistant, as well as for those who require large doses of steroids to sustain remission (steroid dependent). These agents are also beneficial for pediatric patients who experience growth inhibition secondary to chronic use of steroids.12 Cytotoxic agents are effective in inducing remission and the duration of remission tends to be longer than that induced by steroids. In those patients who relapse after cytotoxic therapy, they may respond to steroids better than before.
Cyclophosphamide at 2 mg/kg per day for 10 to 12 weeks given alone or with prednisone (50 to 75 mg/m2) is very effective in inducing remission and restoring steroid responsiveness for patients who were previously steroid dependent and then became steroid resistant. Alternatively, chlorambucil at 0.1 to 0.2 mg/kg per day may be used. This agent, however, is associated with more adverse effects than cyclophosphamide. Azathioprine has also been used; however, treatment for 6 to 12 months is often needed before any favorable response is apparent.
The immunosuppressive effect of cytotoxic agents, with or without the concurrent use of steroids, can result in serious infections, which are the primary cause of death for patients with minimal-change nephropathy. Other toxicities associated with cyclophosphamide include gonadal fibrosis, which results in sterility, hemorrhagic cystitis, alopecia, and a potential to develop malignancy in those on long-term treatment.
Calcineurin Inhibitors
Cyclosporine decreases lymphokine production by activated T lymphocytes and thereby reduces proteinuria by reversing the lymphokine-induced alterations in the anionic charge and permeability of the GBM to albumin. For patients with steroid-sensitive or steroid-dependent disease, cyclosporine induces remission in 80% to 85% of patients. However, the disease-free period is not often sustained, and relapse, which is usually not as responsive to cyclosporine retreatment, may occur as soon as the drug is tapered or discontinued. The steroid-sparing effect of cyclosporine is also useful for steroid-dependent patients, especially those who have experienced significant adverse effects.
Dosage The usual starting dose of cyclosporine for remission induction is 5 mg/kg per day for adults and 100 to 150 mg/m2 per day for children. Similar dosages are used to maintain remission long term. The optimal cyclosporine blood concentrations, as well as the need to monitor them, are controversial. No correlation has been found between the severity of the cyclosporine-induced tubulointerstitial lesions and the mean dose or trough drug concentration. However, monitoring of the area under the serum concentration–time curve has been suggested and target exposures have been proposed.38 Testing the in vitro sensitivity of peripheral blood lymphocytes to cyclosporine in the presence of a T-cell mitogen may offer a novel method to predict response and individualize therapy.39
Adverse Events Adverse events such as rise in serum creatinine, hypertrichosis, and gingival hyperplasia are quite common. Long-term therapy may result in persistent hypertension and progressive renal failure. Cyclosporine should not therefore be given for more than 4 months in the absence of any beneficial effect. Consequently, it is indicated for patients (a) who relapse frequently or are steroid dependent, after failing to respond to a course of cyclophosphamide; (b) for whom cyclophosphamide is contraindicated or when gonadal toxicity is a concern; (c) who are steroid dependent when a “steroid holiday” is needed for catch-up growth and puberty; or (d) who have steroid-resistant disease.36
Levamisole
Levamisole, an immunostimulant, can promote the maturation of young T cells and restore the function of T cells and phagocytes when the immune system is depressed. It may also inhibit the production of an immunosuppressive lymphokine. Levamisole was found to have a steroid-sparing effect and was capable of maintaining remission in children who had frequent relapse steroid-dependent nephrotic syndrome.40 In addition, it is as effective as cyclophosphamide in reducing relapse rate and steroid dosages.41 The adverse effect of levamisole are mild neutropenia, which is generally reversible, and GI upsets. At present the drug is no longer available in the United States; however, it is recommended by the KDIGO guidelines as a steroid-sparing agent.10
Mycophenolate Mofetil
Mycophenolate mofetil is an immunosuppressant that can suppress T- and B-cell lymphocyte proliferation, B-lymphocyte antibody production, and expression of adhesion molecules. It is reported to have steroid-sparing effects and is useful in frequently relapsing, steroid-dependent and steroid-resistant patients, as well as in those who fail cytotoxic therapy.42
Rituximab
Rituximab has been observed to induce long-term remission with repeated doses,43 possibly through a direct effect on the podocyte actin cytoskeleton. However, its use is not recommended by the KDIGO clinical practice guidelines due to the lack of randomized trials and risk for serious adverse effects.10
Prognosis
The long-term prognosis of most patients with minimal-change disease is good. The majority of pediatric patients will not experience any relapse of the disease 10 years after the initial onset, and most will be free of the proteinuria after puberty. In adults, an 85% to 90% survival rate is seen 10 years after disease onset. Although this condition may spontaneously remit in up to 70% of untreated adults, life-threatening complications may be associated with untreated nephrotic syndrome. Significant deterioration in renal function is uncommon in both adult and pediatric patients and is observed only in those who are steroid resistant or steroid dependent. Because of the overall favorable outcome of the disease and the relatively uncommon progression into chronic renal failure, aggressive use of cytotoxic agents is not indicated even for most patients with frequent relapses. Toxicities associated with aggressive therapy do not justify the need to induce remission in those patients who fail to respond to steroids and the nonaggressive use of cytotoxic agents. Symptomatic therapy with diuretics to control edema, in conjunction with a low-salt diet and albumin infusion as needed for acute development of anasarca, is often a more rewarding therapeutic approach. NSAIDs and ACEIs may also be used to reduce the proteinuria.
Focal Segmental Glomerulosclerosis
Etiology and Epidemiology
FSGS is a clinicopathologic condition that can be idiopathic (primary) or secondary to a variety of causes. FSGS accounts for less than 20% of the cases of idiopathic nephrotic syndrome in children and approximately 40% in adults44; however, it may account for 36% to 80% of the cases in African Americans. The incidence of FSGS has been rapidly increasing, so that it now is the most common glomerular disease that ultimately leads to ESRD. Conditions such as sickle cell disease, cyanotic congenital heart disease, and morbid obesity can induce hemodynamic stress on an initially normal nephron population and result in FSGS. Severe glomerular injury can also be seen in patients with nephropathy associated with heroin abuse, human immunodeficiency virus (HIV) infection, and genetic mutations involving the podocin and WT1 genes. A recent case series identified the association of FSGS and proteinuria in bodybuilders after long-term anabolic steroid abuse.45 In addition, heroin, pamidronate, and interferon have been associated with FSGS.44 The primary and secondary sclerotic lesions may be morphologically similar, but they represent diseases with different courses and responses to therapy.
Pathophysiology
Sclerotic lesions are characteristically found in some of the glomeruli (focal) and usually involve only a portion of the glomeruli (segmental).44 Similar to minimal-change disease, fusion of foot processes is commonly seen in those glomeruli that are not sclerotic. It is thought that both minimal-change disease and FSGS share similar pathogenetic mechanisms, with FSGS resulting in severe injury to the glomerular epithelial cells. During the early stage of FSGS, only a small number of glomeruli may have the segmental sclerotic lesion, and the disease may be confined to the juxtamedullary region. If an inadequate number of glomeruli are sampled during renal biopsy, the diagnosis of FSGS may be missed, or the patient may be thought to have minimal-change disease. Resistance to steroid therapy may thus be one of the first clues that the patient, indeed, has FSGS rather than minimal-change disease. Alternatively, a patient may have the steroid-sensitive minimal-change disease initially, which subsequently progresses to steroid-resistant FSGS.
Clinical Presentation
Almost all the patients present with proteinuria, and many of them have all the features of nephrotic syndrome. The proteinuria is nonselective, containing albumin and other higher-molecular-weight proteins, and is usually less severe when compared to patients who have minimal-change disease. Hypertension, microscopic hematuria, and renal dysfunction may be seen in up to half of the patients. Reduced renal function becomes more prevalent as the disease progresses.
The presenting clinical features in nephrotic adults with minimal-change nephropathy can be indistinguishable from that of FSGS, and renal biopsy is therefore critical in the diagnosis of adults with nephrotic syndrome. African Americans have a fourfold higher risk of developing FSGS than white or Asian patients. They tend to develop the disease earlier and present with nephrotic range proteinuria more often. They are less responsive to steroids and are more likely to experience a rapid decline in renal function, resulting in ESRD.
TREATMENT
Pharmacologic Therapy
The treatment of FSGS is controversial because of the lack of data from randomized, prospective, controlled trials.
Steroids
A course of prednisone (1 to 2 mg/kg/day) with tapering after 3 to 6 months of treatment is first used for nephrotic patients.44 Urinary protein excretion and serum albumin concentration should be monitored to assess efficacy. The median time to induce complete remission is 3 to 4 months, although 5 to 9 months may be needed in some patients. In general, 30% to 50% of all patients are expected to be resistant to steroids, after at least 4 months of therapy. Patients with diffuse mesangial IgM deposition may be prone for steroid resistance.46
If the patient develops a relapse after an adequate response to the initial treatment, a second course of steroids is generally sufficient. However, if relapse occurs frequently, cytotoxic agents or cyclosporine would be indicated. For patients who are not nephrotic, their relatively favorable prognosis does not support using steroids or other immunosuppressive agents. However, close follow-up and good blood pressure control with ACEIs are necessary to minimize disease progression.44
Most of the studies conducted thus far include mostly white patients. In a retrospective review of 72 patients that included 65 African American patients, steroid use was not associated with renal survival or the induction of proteinuria remission.47 The initial creatinine level, blood pressure, and severity of renal lesions are significant factors for renal survival. About one third of the patients who received steroids developed complications such as diabetes and significant weight gain.
Cytotoxic Agents
When used with steroids during initial therapy, cytotoxic agents were not found to offer any additional beneficial effect.44,48 Randomized clinical trials are not available to support their use as first-line therapy.10
Calcineurin and Rapamycin Inhibitors
In steroid-resistant patients, cyclosporine therapy has produced a complete or partial remission in 70% of patients, with a relapse rate of 47%.49 Tacrolimus may also be used with similar effects.50 Therapy continued for 12 months before slow tapering is more likely to maintain remission. In patients with diabetes, psychiatric disorder, or severe osteoporosis, calcineurin inhibitor may be a first-line therapy due to the concern for steroid side effects.44 The effect of sirolimus on proteinuria has been found to be conflicting; however, it may cause a rapid decline in GFR, and hence its use for FSGS is not recommended.51
Mycophenolate Mofetil
Mycophenolate mofetil has been reported to have favorable effects for patients who were steroid resistant. After inducing remission with high-dose IV methylprednisolone and oral cyclosporine, a combination of cyclosporine and mycophenolate, followed by mycophenolate alone, can sustain long-term remission, preserve renal function, and improve blood pressure control.52 However, due to the varied experiences from different investigators, further studies are needed to define the role of this agent among the various treatment options.
ACEIs and ARBs
![]() Because of the lack of a consistently effective regimen for primary FSGS, many patients with mild disease are treated conservatively. ACEIs and ARBs are effective in reducing proteinuria and stabilizing renal function in many patients with primary or secondary FSGS. Control of blood pressure and hyperlipidemia are important as well.44 For patients who have nephrotic range proteinuria, an elevated serum creatinine concentration, and interstitial scarring on biopsy, corticosteroids with or without immunosuppressive agents are often used.
Because of the lack of a consistently effective regimen for primary FSGS, many patients with mild disease are treated conservatively. ACEIs and ARBs are effective in reducing proteinuria and stabilizing renal function in many patients with primary or secondary FSGS. Control of blood pressure and hyperlipidemia are important as well.44 For patients who have nephrotic range proteinuria, an elevated serum creatinine concentration, and interstitial scarring on biopsy, corticosteroids with or without immunosuppressive agents are often used.
Prognosis
ESRD develops within 10 years in 10% or less of the 30% to 50% of adults and children who had attained complete remission.49 For those patients who are resistant to therapy, the rate of renal function deterioration to ESRD may be rapid, within 1 year, or slow, over as long as 10 to 20 years; approximately 50% develop ESRD within 10 years. Those patients with severe proteinuria (>10 to 15 g/day), high serum creatinine concentration at diagnosis, initial steroid resistance, or interstitial fibrosis on renal biopsy are likely to have a more rapid decline in renal function. African American patients may also have a higher risk. Kidney transplantation is often indicated for those patients who develop ESRD; however, FSGS has recurred in 40% of the renal allografts soon after transplantation.44 Children, nonblack race, and those with severe disease or rapid progression to ESRD prior to transplantation are more likely to experience a recurrence. The proteinuria may reappear within hours after transplantation, and graft failure may occur in one third to one half of the patients. The median time to recurrence was reported to be 14 days in one study. Although cyclosporine is ineffective in preventing the recurrence of nephrotic syndrome after transplantation, a high dose of the agent (up to 35 mg/kg/day) induces a remission of the recurrent disease. ACEIs and plasmapheresis are also used to prolong graft survival. The effectiveness of these therapies and the rapid recurrence of the disease in the transplanted kidney substantiate the possibility that a circulating humoral mediator is responsible for the nephropathy. Plasmapheresis to remove the mediator was found to be effective in inducing a remission.44
Membranous Nephropathy
Etiology and Epidemiology
Membranous nephropathy is the most common disorder responsible for idiopathic nephrotic syndrome in adults, accounting for about 20% to 25% of cases. It is also a frequent cause of renal failure secondary to glomerulonephritis. The hallmark histologic features of membranous nephropathy are glomerular capillary wall thickening with subepithelial deposits under light and electron microscopy. Autoimmunity is responsible for 70% to 80% of the cases. Autoantibodies toward phospholipase A2 receptor (PLA2R) and neutral endopeptidase (NEP) have been discovered recently.53 The presence of bovine serum albumin (BSA) and anti-BSA antibodies in certain patients suggests that food antigens may be involved in the pathogenesis. Further anti-PLA2R antibodies appear to predict disease activity and response to therapy.53
About 25% of adults and 80% of children have secondary causes.54 In the United States, the most common etiologies are autoimmune diseases (e.g., lupus), infection (e.g., hepatitis B and C), syphilis, neoplasm (e.g., carcinoma of the lung, breast, GI tract, or kidney), and medications (e.g., gold, penicillamine, or captopril). Malaria and schistosomiasis are common causes in other parts of the world. De novo membranous nephropathy can also occur in the allografts of renal transplant patients. Because the responses to therapy as well as the prognosis for idiopathic and secondary membranous nephropathy are different, it is important to identify any potential underlying causes for the nephropathy prior to treatment. Although this glomerular disease can occur at any age, the peak incidence is between ages 30 and 50 years and is especially likely in patients older than age 50 years who present with nephrotic syndrome.54
Pathophysiology
Examination of kidney tissue under light microscopy reveals normal mesangium and normocellularity. The glomerular capillary wall may be thickened in well-developed lesions. In the advanced stage, the epithelial side of the capillary wall is markedly thickened, and intramembranous deposits are found. Progressive changes in capillary lumen patency parallel those in the GBM, resulting in glomerulosclerosis with capillary collapse and tubular atrophy in end-stage membranous nephropathy. Immunofluorescence microscopy shows strong capillary wall staining of IgG and C3 on the epithelial side of the basement membrane. Antibody-mediated immune injury appears to be the main pathogenetic mechanism. The immune complex can be formed in situ or deposited from circulating immune complexes.
Clinical Presentation
Most patients with membranous nephropathy present with heavy proteinuria (exceeding 3.5 g/day). Those patients excreting large amounts of IgG and α1-microglobulin, indicating more significant tubulointerstitial damage, have a lower remission rate, and are more likely to progress toward renal failure.54
The signs and symptoms are usually insidious in onset and may consist of anorexia, malaise, edema, anasarca, or ascites, and pericardial and pleural effusions may also be present. As a result of a hypercoagulable state, pulmonary embolism may develop but rarely results in death. The incidence of renal vein thrombosis varies from 5% to 62%, and membranous nephropathy should be suspected when there is a sudden onset of hematuria, loin pain, pulmonary embolus, fluctuating or worsening proteinuria or GFR, renal tubular acidosis, or an increase in leg edema. Hypertension is found in approximately 30% of patients and is more common with renal insufficiency or in advanced disease.
In addition to heavy proteinuria, urinalysis often reveals lipiduria and oval fat bodies. Microhematuria is seen in fewer than 25% of patients, and gross hematuria and red cell casts are rare. In idiopathic membranous nephropathy, the serum complement concentrations are normal. Low levels of complement should alert one to search for secondary causes, such as lupus, hepatitis B infection, or an alternative diagnosis. Similarly, antinuclear antibodies, anti-DNA antibodies, rheumatoid factor, hepatitis B serologies, and serum cryoglobulins are generally negative in idiopathic membranous nephropathy. Occult malignancy has been found in as many as 10% of elderly patients with membranous nephropathy.
TREATMENT
The treatment of idiopathic membranous nephropathy is controversial and ranges from supportive therapy to immunosuppression. Conservative management of patients with mild disease includes edema control with salt restriction and diuretics5 and reduction of proteinuria with protein restriction and ACEIs (Fig. 32-4).10,55 Management of hypertension and hyperlipidemia is required for most patients, whereas prophylactic anticoagulation, despite having benefits shown to outweigh the risks, is usually given only for patients with renal vein thrombosis or documented pulmonary embolus.34,55
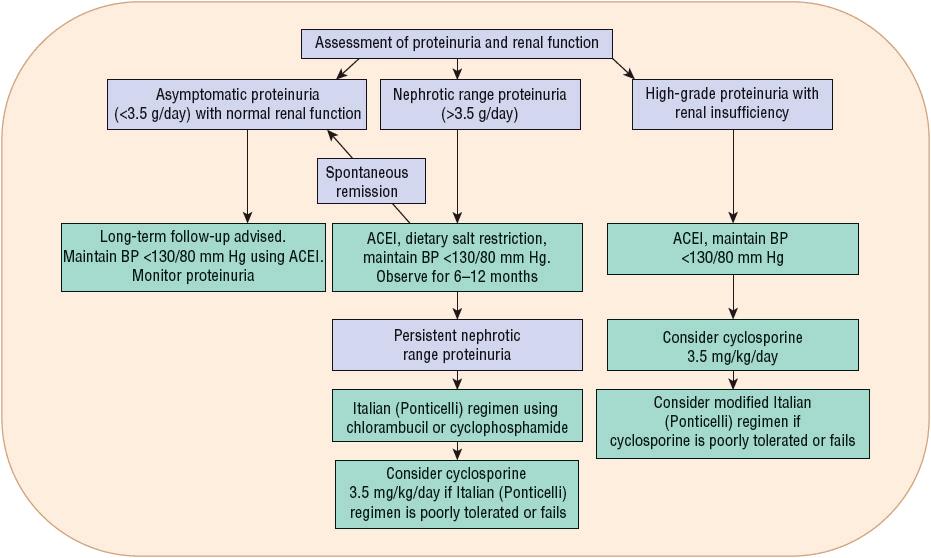
FIGURE 32-4 Treatment algorithm for idiopathic membranous nephropathy. (Adapted from reference 55.)



