Genetics
OBJECTIVES
The reader will be able to:
- Define the terms: allele, dominant allele, genotype, genetic polymorphism, haplotype, homozygous, heterozygous, drug idiosyncrasy, monogenic, pharmacogenetics, pharmacogenomics, phenotype, polymodal frequency distribution, polygenic, poor metabolizers, recessive allele, unimodal frequency distribution.
- Give three examples of inherited variability in pharmacokinetics and three in pharmacodynamics.
- Demonstrate how population studies and studies in twins can be used to indicate the existence of genetic polymorphism.
- State under what circumstances phenotype status is of therapeutic value.
- Describe at least two ways, other than twin studies, in which genetic polymorphism can be demonstrated and list two examples of each.
 nheritance accounts for a large part of both the striking and the subtle differences among individuals, including much of the variation in response to an administered drug. Unlike other sources of variability, inherited traits remain an individual’s characteristic for a lifetime. Our knowledge of this area is expanding very rapidly with the characterization of the human genome. Pharmacogenetics is the study of inherited variations in drug response. Pharmacogenomics is the application of genomic information to the identification of putative drug targets and to the causes of variability in drug response. Before proceeding to consider specific examples, some definitions are important to an understanding of the subject.
nheritance accounts for a large part of both the striking and the subtle differences among individuals, including much of the variation in response to an administered drug. Unlike other sources of variability, inherited traits remain an individual’s characteristic for a lifetime. Our knowledge of this area is expanding very rapidly with the characterization of the human genome. Pharmacogenetics is the study of inherited variations in drug response. Pharmacogenomics is the application of genomic information to the identification of putative drug targets and to the causes of variability in drug response. Before proceeding to consider specific examples, some definitions are important to an understanding of the subject.
The basic biological unit of heredity is the gene. Genotype is the fundamental assortment of genes of an individual, the blueprint, whereas phenotype is the outward characteristic expression of an individual, such as the color of one’s eyes. The mode of inheritance is either monogenic or polygenic, depending on whether it is transmitted by a gene at a single locus or by genes at multiple loci on the chromosomes. Monogenically controlled conditions are often detected as a dramatic and abnormal drug response, that is, a drug idiosyncrasy. They may also be detected in population studies by a polymodal frequency distribution of the characteristic or some measure of it, as depicted for the clearance of Drug D in Fig. 12-6 (Chapter 12, Variability). Polygenically controlled variations tend to have the appearance of a unimodal frequency distribution.
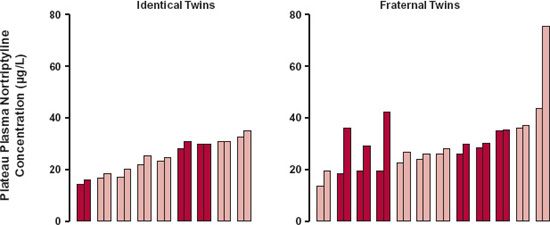
FIGURE 13-1. The much smaller intrapair variability in plateau plasma concentration of nortriptyline between nine identical twins than between 12 fraternal twins indicates that genetics plays a major role in nortriptyline pharmacokinetics. Female (dark color); male (light color). (From: Alexanderson B, Evans DA, Sjöqvist F. Steady-state plasma levels of nortriptyline in twins: influence of genetic factors and drug therapy. Br Med J 1969;4:764–768.)
Historically, the role of genetics in drug response has been demonstrated by showing that interindividual variation in phenotypic behavior in identical twins is much smaller than that between nonidentical, gender-matched twins, or in any two randomly selected age-, gender-, and weight-matched subjects (Fig. 13-1). Although this approach in identifying the relative importance of genetics is helpful, today, more definitive data concerning mechanisms are obtained by genomic profiling using molecular biology techniques.
The human genome comprises over 1.5 million single-nucleotide polymorphisms (SNPs), locations along DNA where a difference exists among individuals in the nucleotide. The alternate forms, each occupying corresponding positions on paired chromosomes, one inherited from the male and the other from the female, are called alleles. Different alleles produce variation in inherited characteristics such as hair color, blood type, or drug metabolizing ability. An allele is dominant if it expresses itself phenotypically and recessive if it does not. An individual possessing a pair of identical alleles is said to be homozygous for the gene and heterozygous when the individual has different alleles for the gene. Both homozygous and heterozygous individuals containing the dominant allele may show the same phenotype, and homozygous individuals with recessive alleles may show another. Alleles are often characterized using the * notation. For example, a gene denoted by *1/*1 is homogenous, and one by *1/*3 is heterogeneous, for allele 1, where allele 1 is often the wild-type, or the most common allele, for the gene found in the population studied. The numbering generally follows the chronologic order in which an allele was identified. Sometimes, it is a set of nearby SNPs on the same chromosome that is inherited as a block, a haplotype, rather than a single SNP, which determines phenotypic behavior.
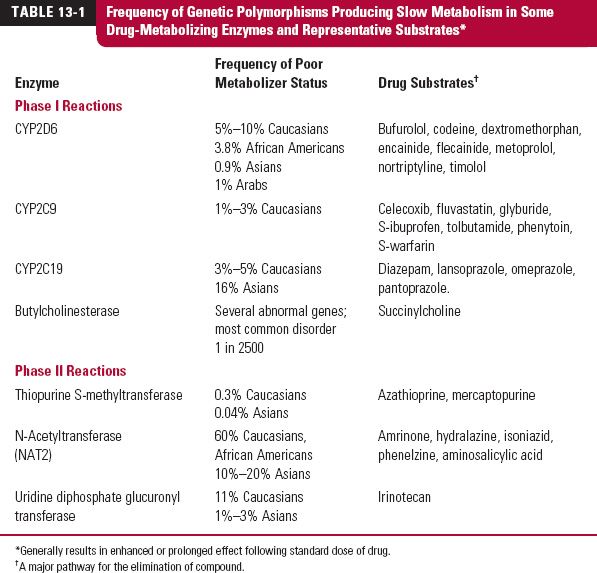
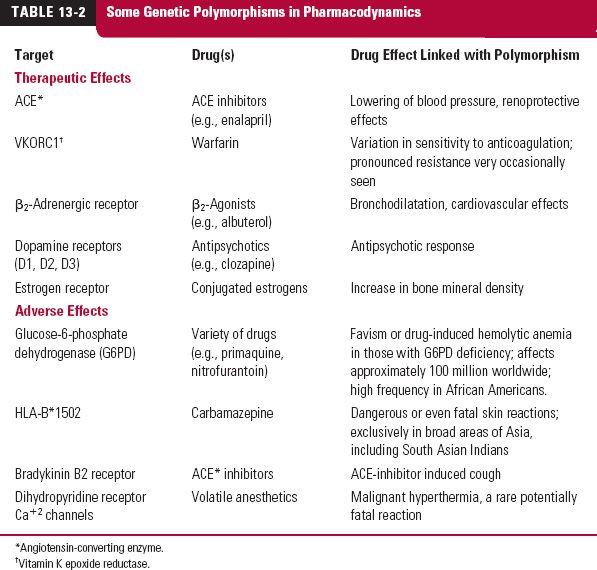
Because the approximately 30,000 genes contain multiple SNPs, identifying the most relevant ones affecting disease susceptibility, drug metabolism and transport, and drug response holds the promise of predicting these for individual patients. Although there are a myriad of possible polymorphisms, historically, a genetic polymorphism has been defined clinically as the condition in which the frequency of the variant occurs in greater than 1% of the population. Tables 13-1 and 13-2 (page 360) list genetic polymorphisms that affect the pharmacokinetics and pharmacodynamics of some drugs. Although focus is on factors determining therapeutic efficacy and adverse effects, the inability of approximately 30% of Caucasians to taste certain drugs such as propylthiouracil, which contains a thiocyanate group, illustrates how genetics also contributes to variability in the senses.
Before considering specific examples, a final general comment is warranted. Commonly, individuals are classified demographically by their ethnicity, such as Caucasians, African American, or Asian. Although, as we shall see, such classification does sometimes help in characterizing differences in response within the population to drugs, it is well to remember that this classification relates essentially to external features, and within each ethnic group, there is often almost as much interindividual variability as between the groups. Clearly, the most important genetic characteristics are those of the individual receiving the drug.
INHERITED VARIATION IN PHARMACOKINETICS
Examples of inherited variability in pharmacokinetics have been almost exclusively restricted to drug metabolism. Comparatively, there is little clinically significant genetic polymorphism currently identified in drug absorption, distribution, and renal or biliary excretion. Nonetheless, genetic polymorphisms have been identified in drug transporters, such as the hepatic uptake transporter, organic anion-transporting polypeptide 1B1 (OATP1B1), and it is likely that clinically important changes in pharmacokinetics will be found especially for those substrates, such as pravastatin, for which hepatic uptake rate limits clearance. When polymorphism in a transporter affects the distribution into a target organ, such as liver or brain, or a site of potential toxicity, modifying the ratio of local unbound drug or metabolite to that in plasma, the effect would be defined as an inherited variation in pharmacodynamics, in efficacy or safety, even though the cause is a pharmacokinetic one. Clearly, in the absence of knowing the unbound concentration at the active site, which is rarely determined clinically, the mechanism for such variability in response cannot be ascribed with confidence.
Several genetic polymorphisms of drug metabolism have now been identified, primarily involving oxidation, but also S-methylation, acetylation, and hydrolysis (see Table 13-1). Most were initially detected by adverse reactions occurring in a distinct group within the population termed, poor metabolizers, following normal doses of the archetypic drugs. Some examples follow.
OXIDATION
Debrisoquine, an antihypertensive agent no longer in use, was the first drug shown to exhibit genetic polymorphism in oxidation. There is a deficiency in the metabolism of this drug in 5% to 10% of Caucasians, poor metabolizers, with wide differences in frequency in other ethnic groups (see Table 13-1). It is a recessive trait, caused by a specific variant of cytochrome P450 2D6 (CYP2D6). Of the remainder of the population, often referred to as extensive metabolizers, some are ultrafast metabolizers, because they possess up to 13 copies of the normal gene within the population, commonly referred to as the wildtype, which is clearly expressed phenotypically in a strong inverse correlation between the number of copies and the area under the curve (AUC) for nortriptyline, which predominantly cleared via CYP2D6 oxidation (Fig. 13-2). This variation in the metabolizing enzyme(s) is the predominant reason for the widespread steady-state plasma nortriptyline concentrations among patients receiving the same dosage regimen of nortriptyline (Fig. 13-2). It is also expressed in the large difference in metoprolol profiles between poor and extensive metabolizers (Fig. 13-3). Recall from Chapter 5, Elimination, although comprising only approximately 2% of the total cytochrome P450 (CYP450) content in the liver, CYP2D6 is responsible for the metabolism of approximately 25% of prescribed drugs, particularly basic compounds, including the β-blocker bufurolol, and the antidepressants, doxepin and paroxetine. With all these drugs, CYP2D6-catalyzed metabolism is the major pathway for elimination.
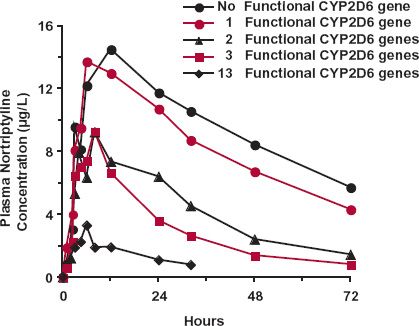
FIGURE 13-2. Strong genetic influence in the pharmacokinetics of nortriptyline is clearly demonstrated by the high correlation between the plasma concentration–time profile and the number of functional CYP2D6 genes possessed by an individual; the larger the number of functional genes, the higher is the clearance and the lower is the exposure profile following a single 25-mg dose of nortriptyline. (From: Dalén P, Dahl ML, Bernal Ruiz ML, et al. 10-Hydroxylation of nortriptyline in white persons with 0, 1, 2, 3, and 13 functional CYP2D6 genes. Clin Pharmacol Ther 1998;63:444–452.)
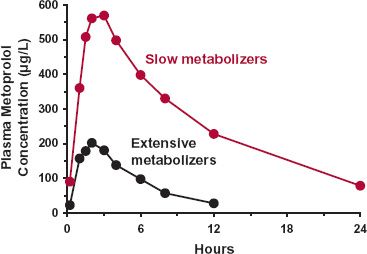
FIGURE 13-3. Plasma metoprolol concentrations after a single oral dose of 200-mg metoprolol tartrate were much higher in poor (colored line) than in extensive (black line) CYP2D6 metabolizers. Because metoprolol is a drug of high hepatic clearance, the difference between poor and extensive metabolizers is expressed in the large difference in oral bioavailability, because of differences in first-pass hepatic loss. (From: Lennard MS, Silas JH, Freestone S, et al. Oxidative phenotype—a major determinant of metoprolol metabolism and response. Reprinted by permission of New Eng J Med 1982;307:1558–1560.)
Genetic polymorphism is not restricted to CYP2D6. Although the wide variation in the daily maintenance dose of warfarin has been known for many years, only recently has it become clear that a significant contribution to this variability is mutation of the cytochrome P450 2C9 (CYP2C9) gene, primarily responsible for the metabolism of the more potent S-isomer of this racemic drug (Fig. 13-4). Other drugs that are predominantly metabolized by CYP2C9, and for which clearance is reduced between 3- to 10-fold of the wild type in poor metabolizers, include tolbutamide, glyburide, and phenytoin.
Many drugs are oxidized with widely differing efficiencies by the different forms of CYP450 in addition to CYP2D6 and CYP2C9, such as cytochrome P450 2C19 (CYP2C19) (see Table 13-1
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


