Genetic Pathology
GENERAL PRINCIPLES
What is the normal human chromosomal complement?
46,XX or 46,XY
What is the normal human haploid number?
23
What is aneuploidy?
Possessing a chromosome number that is not a multiple of the normal haploid number, typically arising from an error in meiosis or mitosis
What are the most common causes of aneuploidy?
Meiotic nondisjunction or failure of chromosomal separation during anaphase of meiosis I (anaphase lag)
What are the most common aneuploidies?
Trisomy and monosomy (Trisomy 21 is the most common chromosomal disorder.)
What is the condition of having one or more additional sets of the haploid number of chromosomes?
Polyploidy (eg, tetraploidy or triploidy)
What is the term for a specific copy of a gene?
Allele
What is the term for loss of part of a chromosome?
Deletion
What is an inversion?
Chromosomal rearrangement resulting from two break points on the same chromosome, with subsequent reversal and reincorporation into the chromosome.
What is the term for two nonhomologous chromosomes exchanging segments?
Translocation
What is a Robertsonian translocation?
An exchange of chromosomal segments between two acrocentric chromosomes, typically resulting in one large and one small chromosome. The small chromosome is often lost.
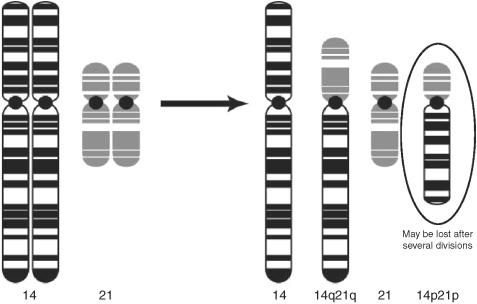
Figure 3.1 Robertsonian translocation.
What type of chromosome has either two short arms or two long arms due to faulty separation of the centromere during meiosis?
Isochromosome
What is the process by which all but one X chromosome in each cell is randomly inactivated early in embryonic development?
Lyonization
What does lyonization produce?
Barr bodies
What is genetic mosaicism?
An individual’s cells are a mixture of differing genotypes (eg, some cells are normal while others have trisomy 21, or XO, or show variable lyonization. Lyonization is a normally occurring mosaicism in which approximately half of a woman’s cells have maternal X inactivation while the remaining cells have paternal X inactivation.
CHROMOSOMAL ABNORMALITIES
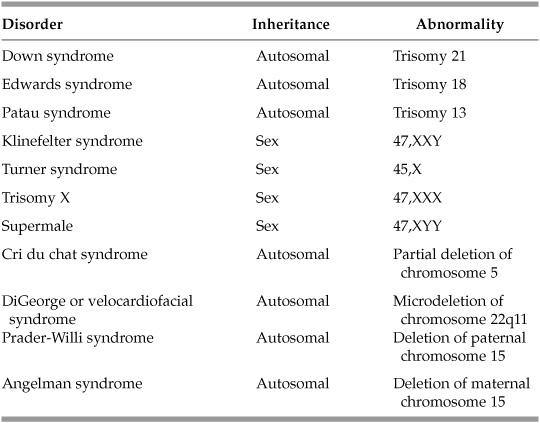
What is the major cause of Down syndrome?
Trisomy 21, usually from maternal meiotic nondisjunction
What are the clinical features of Down syndrome?
Flat nasal bridge, low-set ears, upslanting epicanthal folds, wide-set eyes, simian crease, endocardial cushion cardiac malformations (including ASD, VSD), esophageal atresia, and mental retardation
What is the name given to another characteristic of Down syndrome in which there are small white spots around the border of the iris?
Brushfield spots
Down syndrome patients are at increased risk of developing which conditions later in life?
Early onset Alzheimer disease and both acute lymphoblastic and myeloid leukemias
What is Edwards syndrome and what are the clinical characteristics?
Trisomy 18—newborn with mental retardation, rocker-bottom feet, micrognathia (small lower jaw), prominent occiput, and congenital cardiac malformations
What is Patau syndrome and what are the clinical characteristics?
Trisomy 13—newborn with mental retardation, deafness, microphthalmia, microcephaly, cleft lip and palate, polydactyly cardiac defects, clenched fists, rocker-bottom feet, and holoprosencephaly
What is the most common cause of a 47,XXY karyotype?
Maternal meiotic nondisjunction
What is the name and clinical phenotype observed in individuals with 47,XXY genotype?
Klinefelter syndrome—tall-statured male with hypogonadism, mild gynecomastia, infertility, and a Barr body seen on buccal smear preparation
Are males with a 47,XYY karyotype easily distinguished from males with a normal 46,XY karyotype?
No, but they are generally tall-with an increased risk of having learning difficulties and/or behavioral problems.
What is the karyotype and common phenotype seen in Turner syndrome?
45,XO—short-statured female, webbed neck, widely spaced nipples, delayed puberty
What cardiovascular anomaly is most often seen with this syndrome?
Coarctation of the aorta
What test could be performed to conclusively diagnose this syndrome?
Karyotype
What is the common phenotype seen in trisomy X (47,XXX)?
Mild mental retardation, menstrual irregularities, and two Barr bodies
What causes a condition characterized by a weak, cat-like cry, severe mental retardation, microcephaly, and congenital heart defects?
Partial deletion of the short arm of chromosome 5 (cri du chat syndrome)
What genetic defect results in DiGeorge syndrome or velocardiofacial syndrome?
Microdeletion of chromosome 22qll
What shared clinical features are associated with microdeletion of chromosome 22q11?
Craniofacial anomalies including cleft palate, congenital heart defects, mental retardation, and increased risk of schizophrenia
What unique features may be used to distinguish DiGeorge syndrome from velocardiofacial syndrome?
Thymic and parathyroid hypoplasia with resultant variable degrees of T-cell immunodeficiency and hypocalcemia
What is genomic imprinting?
An epigenetic process related to differences in DNA methylation of an allele inherited from the mother or the father. DNA methylation results in inactivation of a gene or possibly an entire chromosome.
What two syndromes are associated with genetic imprinting of the long arm of chromosome 15?
- Prader-Willi
- Angelman syndrome
Which syndrome is associated with the deletion of a normally active paternal allele of chromosome 15 and what are the clinical features?
Prader-Willi (P for paternal)—mental retardation, truncal obesity, hypogonadism, and small hands and feet
Which syndrome is associated with the deletion of a normally active maternal allele of chromosome 15 and what are the clinical features?
AngelMan’s syndrome (M for maternal)—mental retardation, mutism, seizures, ataxia, inappropriate laughter
INHERITANCE PATTERNS
What is the term for having two identical alleles for a given gene?
Homozygous
What is the term for having two different alleles for a given gene?
Heterozygous
What is the term for having only one copy (allele) of a gene?
Hemizygous
What is a point mutation?
Mutation or changing of a single nucleotide
What is a frameshift mutation?
Deletion or insertion of one or more nucleotides that is not a multiple of three, therefore changing the transcriptional reading frame
Point mutation that causes substitution of one amino acid for another in the protein sequence. It can be conservative (has no effect on protein function) or nonconservative (usually affects protein function deleteriously).
What is a nonsense mutation?
Point mutation that changes the encoded amino acid to a stop codon and causes formation of a truncated version of the protein. It is almost always deleterious.
What is a silent mutation?
Point mutation in which the single base change does not code for a different amino acid, so no disease condition arises
What is a trinucleotide repeat mutation?
Expansion of a repeated sequence of three nucleotides
What is the term describing the tendency for a disorder to increase in severity or appear at an earlier age as it is passed on to the next generation?
Anticipation—often seen with trinucleotide repeat mutations
A disorder in which only one copy of the mutant gene is necessary to cause disease is what type of inheritance?
Autosomal dominant (AD)
A disorder in which two copies of a mutant gene are necessary to cause disease is what type of inheritance?
Autosomal recessive (AR)
What pattern of inheritance causes disorders usually in males and creates heterozygous female carriers due to a mutant gene on the X chromosome?
X-linked recessive
What is co-dominance?
Two alleles share dominance (eg, AB blood group)
What is variable expressivity?
Identical genotypes display a range of phenotypic manifestations and/or severities
What is incomplete penetrance?
Not all individuals with a mutant genotype show the mutant phenotype
What type of inherited genetic disorder is the result of the combined action of the alleles of more than one gene and usually has more complex hereditary patterns?
Polygenic disorder
What are some examples of polygenic disorders?
Obesity, atherosclerosis, alcoholism, autism, schizophrenia, baldness, cleft palate, idiopathic gout, diabetes mellitus, high blood pressure
AUTOSOMAL DOMINANT DISORDERS
What AD connective tissue disease is caused by a mutation in fibrillin?
Marfan syndrome
What are the hallmark defects of this disease?
- Skeletal—tall, thin, arachnodactyly
- Ocular—ectopia lentis (dislocation of ocular lens)
- Cardiovascular—aortic dilation leading to aortic aneurysm or dissection; mitral valve prolapse
A woman with Marfan syndrome and her normal husband wish to have children. What is the probability that their child will have Marfan syndrome?
50%
What is the genetic mutation and the pattern of inheritance for Huntington disease?
AD inheritance of a trinucleotide repeat (CAG) on chromosome 4
What is the physical cause of Huntington disease?
Atrophy of caudate nuclei, putamen, and frontal cortex
What are the resulting symptoms and at what age do they commonly manifest?
Huntington disease typically presents later in life (40-50 years) with involuntary erratic movements (choreiform or extrapyramidal movements) and slow but progressive cognitive decline.
What molecular genetic tool may be used for early diagnosis of this disease?
PCR or DNA sequencing; previously restriction fragment length polymorphism (RFLP) studies were used
What is the explanation for the observation of anticipation, especially associated with paternal transmission?
A progressive expansion of the trinucleotide repeat from generation to generation, which is more likely occur amongst the vast number of sperm produced by the father than the small number of ova produced by the mother.
What AD disease is caused by a mutation of the low-density lipoprotein (LDL) receptor resulting in high serum levels of LDL cholesterol and early onset on atherosclerosis?
Familial hypercholesterolemia
What are the other characteristics of this disease involving deposits of cholesterol in certain areas of the body?
- Xanthomas—deposits in skin and tendons
- Arcus corneae—deposits around periphery of cornea
What AD disorder consists of telangiectases (dilated capillaries) of the skin and mucous membranes with periodic bleeding ranging from epistaxis (nosebleeds) to gastrointestinal hemorrhage?
Hereditary hemorrhagic telangiectasia, otherwise known as Osler-Weber-Rendu syndrome
What AD disorder is characterized by bilateral destruction of renal parenchyma due to multiple expanding cysts, ultimately leading to renal failure?
Autosomal dominant polycystic kidney disease (ADPKD)
In the AD disorder hereditary spherocytosis, what causes the characteristic spheroidal erythrocytes?
Defects of the erythrocyte membrane proteins, most commonly spectrin
What test is used to diagnosis hereditary spherocytosis?
Osmotic fragility test
What type of anemia often results from defective erythrocytes?
Hemolytic anemia due to trapping and destruction of the erythrocytes in the spleen
What are the phakomatoses (“neurocutaneous syndromes”)?
Neurofibromatosis (NF) 1 and 2, tuberous sclerosis, von Hippel-Lindau disease (VHL), Sturge-Weber syndrome, and ataxia telangiectasia
Which phakomatoses are hereditary and what are the inheritance patterns?
- Autosomal dominant—NF1 and NF2, tuberous sclerosis, VHL
- Autosomal recessive—ataxia telangiectasia
- Nonhereditary/sporadic—Sturge-Weber syndrome
What are the hallmarks of neurof ibromatosis 1 (NF1) or von Recklinghausen disease?
Multiple neurofibromas (tumors) anywhere on the body
- Café au lait spots (pigmented skin lesions)
- Lisch nodules—pigmented iris hamartomas
- Skeletal lesions—scoliosis, bone cysts
- Greater risk of developing other tumors—Wilms tumors, pheochromocytoma
What is the hallmark of NF2?
Bilateral acoustic neuromas
What are the clinical features of tuberous sclerosis (incomplete penetrance, variable expressivity)?
Cerebral hamartomas (tubers); renal angiomyolipomas and cysts; hypopigmented skin macules (“ash-leaf spots”); mental retardation; seizures
What are the characteristics of von Hippel-Lindau (VHL) disease?
Capillary hemangioblastoma (or cavernous hemangioma) of cerebellum, retina, or sometimes brain stem and spinal cord; cysts in pancreas, liver, and kidneys; propensity to develop renal cell carcinoma
A mutation in what type of gene is the cause of von Hippel-Lindau disease?
Tumor-suppressor gene (specifically the VHL gene)
On which chromosome is this gene located?
Chromosome 3 (short arm)
What AD disease is the most common growth plate disorder and a major cause of dwarf ism?
Achondroplasia
What are the hallmarks of achondroplasia?
Skeletal abnormalities; shortened arms and legs, but relatively normal-length trunk; large head with protruding forehead and depression at the root of the nose
What diseases are caused by trinucleotide repeat expansion?
Huntington disease (AD), myotonic dystrophy (AD), Friedreich ataxia (AR), fragile X syndrome (X-linked)
What are the features of myotonic dystrophy?
Muscular dystrophy and myotonia; cataracts; hypogonadism; frontal balding
The finding of bilateral leukocoria (white reflection) when checking for a red reflex on an infant screening exam is concerning for what disease process?
Retinoblastoma
Would this type of retinoblastoma be hereditary (familial) or sporadic?
Hereditary (AD), because it is present bilaterally.
What is the two-hit hypothesis?
The concept that both alleles of a gene must be mutated for a cancer to develop. If the first mutation is inherited (as in hereditary retinoblastoma) the likelihood of developing cancer is increased as only one sporadic mutation is needed to acquire “two-hits.”
What gene is mutated in retinoblastoma?
Retinoblastoma 1 (RBI), a tumor-suppressor gene on chromosome 13
Is a child with bilateral retinoblastoma at an increased risk for other cancers later in life?
Yes, because the mutated RBI gene is present in other tissues, which may subsequently acquire sporadic mutations.
What primarily AD group of disorders is the result of a mutation in type I collagen?
Osteogenesis imperfecta (OI)
What are the hallmarks of this disease?
Brittle bones often resulting in fractures, easy bruising, blue sclera in some types, hearing loss, and dentinogenesis imperfecta in some types
Which type of OI is the result of a null allele, and which types of OI are the results of missense mutations?
- OI type I—null allele
- OI types II, III, IV—missense mutations
AUTOSOMAL RECESSIVE DISORDERS
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


