- what genetic epidemiology is;
- some of the major recent advances in genetics;
- the major methods for estimating heritability;
- the difference between monogenic and complex disease;
- the major gene mapping strategies;
- how genetic epidemiology can reveal unknown disease pathways;
- when it is appropriate to utilize genetic testing;
- the problems associated with genomic profiling and direct to consumer genetic testing.
What is genetic epidemiology?
Genetic epidemiology is the study of the role of genetic factors in determining health and disease in families and in populations, and the interplay of these genetic factors with the environment. Early in the field’s history, investigators were primarily concerned with estimating the relative importance of genes and environment in disease aetiology. Today, the explosion of molecular genetic technology has propelled the field swiftly forward, so that the focus is now on identifying the actual functional genetic variants that predispose to disease. These advances are slowly starting to filter through to clinical practice. Genetic testing has been used increasingly over the past two decades to assist in the screening, carrier testing and/or diagnosis of various conditions. The continued pace of technological advances promises a future in which an individual may be able to access their entire genetic sequence should they wish to, and to understand its implications. In parallel, population and case-based epidemiological studies will continue to reveal mechanisms and pathways underlying disease aetiology, which will become the drug targets of tomorrow.
This chapter outlines the scope of genetic epidemiology as well as some of the major advances that have occurred in the field over the last few years. Genetics contains a plethora of terms which may be unfamiliar to clinicians and students of epidemiology. Whilst a complete treatise on genetic nomenclature is beyond the scope of this text, we have provided the reader with a glossary of terminology that might prove useful. The reader is referred to any of the classic texts in genetics for a fuller explanation of these terms and basic genetics in general (see also the further reading at the end of this chapter and the glossary sections at the end of the book).
What’s new in genetics?
A high quality ‘finished’ sequence of the human genome was completed in 2003 to much fanfare. The Human Genome Project was the culmination of thirteen years’ painstaking effort from scientists across the globe and millions of dollars of public and private funding. The end result was the ~3 billion base pair genetic sequence of a single reference individual. The importance of the human genetic sequence cannot be overemphasised, but essentially revolves around the simple premise that if the human genetic sequence is known, then its function can begin to be understood.
The focus then shifted to understanding variation in the sequence between different individuals. The human genome sequence is almost exactly the same in all people (i.e. 99.9%), but variation in this sequence between different individuals helps explain why some people are more susceptible to disease than others. The SNP Consortium was established in 1999 to discover and catalogue point mutations in the human genetic sequence called single nucleotide polymorphisms (SNPs) which are thought to occur roughly 1 to every 300 base pairs of sequence. Ten years on, there are well over 20 million SNPs in the database, and the focus has again shifted to how best to use this information for the mapping of human disease.
The International HapMap Project and its successor the 1000 Genomes Project have continued SNP discovery efforts but have also focused on documenting the correlation between these genetic markers. Markers in close physical proximity to each other on the genome also tend to be correlated with each other, a phenomenon known as linkage disequilibrium. This means that if an individual has one allele at a particular locus, they are more likely to have a particular allele at an adjacent locus. The significance is that if the correlation between markers is known, then it is possible to genotype a subsample of markers across the genome (rather than all of them) and obtain approximately the same amount of genetic information, but at far reduced cost.
Monogenic versus polygenic diseases and traits
Monogenic diseases or Mendelian diseases are predominantly the result of a single gene. In other words, if an individual has a copy of the risk allele (in the case of a dominant disease/phenotype), or the risk genotype (in the case of a recessive disease/phenotype) then they have a high probability of developing the disease. Monogenic diseases are typically transmitted through pedigrees according to simple Mendelian principles. Examples include cystic fibrosis, phenylketonuria, Huntington’s Chorea, and sickle cell anaemia. Mendelian diseases (and their associated genetic variants) tend to be rare in populations because of the action of natural selection. That is, individuals who have the disease are often at a disadvantage in terms of survival and or reproduction and are therefore less likely to reproduce and hence pass deleterious variants on to their offspring. Thus the genetic variants that underlie Mendelian traits and diseases tend to be uncommon at the population level, especially diseases transmitted through a dominant mode of inheritance.
Polygenic diseases, as the name suggests, are caused by the combined action of many genes of small effect plus environmental influences. Polygenic diseases are sometimes referred to as complex diseases or common diseases. These diseases provide the biggest financial burden to society because of their high prevalence. Examples include asthma, coronary heart disease, hypertension, and types 1 and 2 diabetes. The genetic basis of these conditions is still being determined, but is likely to involve many common variants of small effect and potentially many as yet undiscovered rare variants of small effect also.
Whilst one doesn’t think of contagious diseases as being polygenic, many infectious diseases also have a genetic component in that some individuals are more genetically susceptible to the disease than others. A classic example involves the CCR5 chemokine receptor gene mutation and susceptibility to infection with the Human Immunodeficiency Virus (HIV). Individuals who carry two copies of the CCR5 Δ32 deletion at the locus have nonfunctioning CCR5 receptors. Since some strains of HIV use the CCR5 protein as a co-receptor to gain entry into cells, individuals who are homozygous for the mutation have strong resistance against these varieties of HIV.
Sometimes the distinction between monogenic diseases and complex diseases is not clear cut. Some complex diseases may contain a small proportion of individuals who are affected with the disease primarily because of the action of a single major gene. These forms of disease behave more like monogenic conditions (i.e. the disease often runs in families with a clear pattern of inheritance). For example, the majority of breast cancers are due to environmental factors of unknown aetiology. However, a small proportion of breast cancer cases are due to autosomal dominant mutations in the BRCA1 and BRCA2 genes. Both of these genes play an important role in maintaining genomic stability by facilitating the repair of double strand DNA breaks. Women who have deleterious mutations in either of these genes have substantially higher risk of breast and ovarian cancer (particularly early onset cancers) and of recurrent primary tumours. Hence families that carry these mutations tend to have many affected individuals, including males too.
Twin studies, adoption studies and migrant studies
Early investigations in genetic epidemiology were focused on determining the relative importance of genetic, shared environmental and unique environmental influences in the aetiology of complex traits and disease. The Classical Twin Design compares the similarity between monozygotic (identical) twins and the similarity between dizygotic (nonidentical) twins. The rationale is that since monozygotic twins share all their genes in common, whereas dizygotic twins share on average half their genes (i.e. the same as ordinary siblings), any excess similarity of monozygotic twins over dizygotic twins must be the result of genetic factors. The classical twin design enables investigators to estimate the proportion of variance in a trait due to genetic factors. This is called the heritability of a trait (Note, the heritability of a trait is actually usually defined as the proportion of variance explained by ‘additive’ genetic factors but this distinction is beyond the scope of this book).
Studies using the classical twin design have suggested that the vast majority of human traits and diseases are influenced by genetic factors to at least some extent. Table 6.1 shows the heritability of some common diseases as assessed using the Classical Twin Design. The implication is that a family history of disease or the presence of a first degree relative with disease is a potential risk factor for developing that condition. The increase in risk of disease will be proportionally greater for diseases that have high heritabilities.
Table 6.1 Heritability of some common diseases.
| High heritability (>70%) | Moderate heritability (>30% and <70%) | Low heritability (<30%) |
| Type I diabetes | Coronary heart disease | Lung cancer |
| Schizophrenia | Rheumatoid arthritis | Breast cancer |
| Alzheimer’s disease | Anorexia nervosa | |
| Bipolar disorder | ||
| Ankylosing spondylitis |
The degree of similarity between family members may also be quantified by the relative risk (λR). This measure is different to the risk ratio (relative risk) defined in Chapter 2. The relative risk here is the risk that a relative ‘R’ of an affected proband will be affected with disease divided by the risk of disease in the general population. For example, it is common to estimate the sibling relative risk (λS), and the parent-offspring relative risk (λPO). If the relative risk equals one (i.e. no difference in risk between related and unrelated individuals), then it is unlikely that there is a genetic component to the condition. A weakness of the relative risk, is that whilst it provides evidence of familiality for a disease, it cannot be used to differentiate between genetic and common environmental sources of similarity between related individuals.
Adoption studies can also be used to estimate the relative contribution of shared environmental and unique environmental influences to a trait of interest. The rationale is that similarity between an individual and their adopted relatives can only be due to shared environment (since these individuals are genetically unrelated). Therefore it is possible to compare the phenotypic similarity between biological relatives and adopted relatives and so estimate the relative importance of shared and nonshared environmental components.
Migration studies compare the risk of disease between individuals in their native country, the risk amongst individuals who have migrated to a new country, the risk amongst second generation migrants and risks amongst the indigenous population. If the risk of disease among migrants and second generation migrants is the same as in their native country, but different from the indigenous group of the country they have migrated to, then this is strong evidence for a genetic component underlying disease.
Genetic mapping of diseases
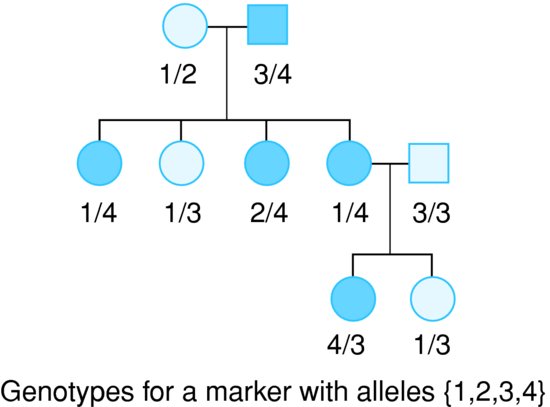
Linkage analysis examines the co-segregation of genetic markers with a disease or trait of interest in pedigrees of related individuals. Consider the pedigree in Figure 6.1. All of the individuals in the family have been genotyped at a single marker of interest. The fact that the ‘4’ allele appears to be transmitted along with the disease provides evidence that there is a disease causing variant on the chromosome somewhere close to the genotyped marker.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


