Chapter 8 General pharmacology
• Qualitative aspects: receptors, enzymes, selectivity.
• Quantitative aspects: dose response, potency, therapeutic efficacy, tolerance.
• Time course of drug concentration: drug passage across cell membranes; order of reaction; plasma half-life and steady-state concentration; therapeutic drug monitoring.
• Individual processes: absorption, distribution, metabolism, elimination.
• Drug dosage: dosing schedules.
• Chronic pharmacology: the consequences of prolonged drug administration and drug discontinuation syndromes.
Individual or biological variation
Pharmacodynamics
Qualitative aspects
Mechanisms
• Ligand-gated ion channels, i.e. receptors coupled directly to membrane ion channels; neurotransmitters act on such receptors in the postsynaptic membrane of a nerve or muscle cell and give a response within milliseconds.
• G-protein-coupled receptor systems, i.e. receptors bound to the cell membrane and coupled to intracellular effector systems by a G-protein. For instance, catecholamines (the first messenger) activate β-adrenoceptors through a coupled G-protein system. This increases the activity of intracellular adenylyl cyclase, increasing the rate of formation of cyclic AMP (the second messenger), a modulator of the activity of several enzyme systems that cause the cell to act. The process takes seconds.
• Protein kinase receptors, so called because the structure incorporates a protein kinase, are targets for peptide hormones involved in the control of cell growth and differentiation, and the release of inflammatory mediators over a course of hours.
• Cytosolic (nuclear) receptors, i.e. within the cell itself, regulate DNA transcription and, thereby, protein synthesis, e.g. by steroid and thyroid hormones, a process that takes hours or days.
Drugs also act on processes within or near the cell by:
• Enzyme inhibition, e.g. platelet cyclo-oxygenase by aspirin, cholinesterase by pyridostigmine, xanthine oxidase by allopurinol.
• Inhibition or induction of transporter processes that carry substances into, across and out of cells, e.g. blockade of anion transport in the renal tubule cell by probenecid is used to protect against the nephrotoxic effects of cidofovir (used for cytomegalovirus retinitis).
• Incorporation into larger molecules, e.g. 5-fluorouracil, an anticancer drug, is incorporated into messenger RNA in place of uracil.
• In the case of successful antimicrobial agents, altering metabolic processes unique to microorganisms, e.g. penicillin interferes with formation of the bacterial cell wall; or by showing enormous quantitative differences in affecting a process common to both humans and microbes, e.g. inhibition of folic acid synthesis by trimethoprim.
Outside the cell drugs act by:
• Direct chemical interaction, e.g. chelating agents, antacids.
• Osmosis, as with purgatives, e.g. magnesium sulphate, and diuretics, e.g. mannitol, which are active because neither they nor the water in which they are dissolved is absorbed by the cells lining the gut and kidney tubules respectively.
Receptors
Radioligand binding studies have shown that the receptor numbers do not remain constant but change according to circumstances. When tissues are continuously exposed to an agonist, the number of receptors decreases (down-regulation) and this may be a cause of tachyphylaxis (loss of efficacy with frequently repeated doses), e.g. in asthmatics who use adrenoceptor agonist bronchodilators excessively. Prolonged contact with an antagonist leads to formation of new receptors (up-regulation). Indeed, one explanation for the worsening of angina pectoris or cardiac ventricular arrhythmia in some patients following abrupt withdrawal of a β-adrenoceptor blocker is that normal concentrations of circulating catecholamines now have access to an increased (up-regulated) population of β-adrenoceptors (see Chronic pharmacology, p. 98).
Enzymes
Irreversible inhibition occurs with organophosphorus insecticides and chemical warfare agents (see Chap. 10), which combine covalently with the active site of acetylcholinesterase; recovery of cholinesterase activity depends on the formation of new enzyme. Covalent binding of aspirin to cyclo-oxygenase (COX) inhibits the enzyme in platelets for their entire lifespan because platelets have no system for synthesising new protein; this is why low doses of aspirin are sufficient for antiplatelet action.
Selectivity
Modification of drug structure
Many drugs have in their design a structural similarity to some natural constituent of the body, e.g. a neurotransmitter, a hormone, a substrate for an enzyme; replacing or competing with that natural constituent achieves selectivity of action. Enormous scientific effort and expertise go into the synthesis and testing of analogues of natural substances in order to create drugs capable of obtaining a specified effect, and that alone (see Therapeutic index, below). The approach is the basis of modern drug design and it has led to the production of adrenoceptor antagonists, histamine receptor antagonists and many other important medicines.
Drug molecules are three-dimensional and many drugs contain one or more asymmetrical or chiral1 centres in their structures, i.e. a single drug can be, in effect, a mixture of two non-identical mirror images (like a mixture of left- and right-handed gloves). The two forms, which are known as enantiomorphs, can exhibit very different pharmacodynamic, pharmacokinetic and toxicological properties.
For example, (1) the S form of warfarin is four times more active than the R form,2 (2) the peak plasma concentration of S fenoprofen is four times that of R fenoprofen after oral administration of RS fenoprofen, and (3) the S, but not the R, enantiomorph of thalidomide is metabolised to primary toxins.
Quantitative aspects
Dose–response relationships
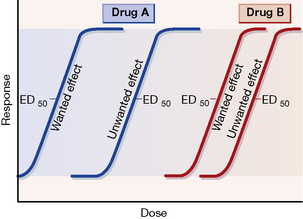
Dose–response curves for wanted and unwanted effects can illustrate and quantify selective and non-selective drug action (see Fig. 8.1).
Potency and efficacy
Ehrlich (see p. 162) introduced the concept of the therapeutic index or ratio as the maximum tolerated dose divided by the minimum curative dose, but the index is never calculated thus as such single doses cannot be determined accurately in humans. More realistically, a dose that has some unwanted effect in 50% of humans, e.g. in the case of an adrenoceptor agonist bronchodilator a specified increase in heart rate, is compared with that which is therapeutic in 50% (ED50), e.g. a specified decrease in airways resistance.
In practice, such information is not available for many drugs but the therapeutic index does embody a concept that is fundamental in comparing the usefulness of one drug with another, namely, safety in relation to efficacy. Figure 8.1 expresses the concept diagrammatically.
Tolerance
Continuous or repeated administration of a drug is often accompanied by a gradual diminution of the effect it produces. A state of tolerance exists when it becomes necessary to increase the dose of a drug to get an effect previously obtained with a smaller dose, i.e. reduced sensitivity. By contrast, the term tachyphylaxis describes the phenomenon of progressive lessening of effect (refractoriness) in response to frequently administered doses (see Receptors, p. 75); it tends to develop more rapidly than tolerance.
Accelerated metabolism by enzyme induction (see p. 93) also leads to tolerance, as experience shows with alcohol, taken regularly as opposed to sporadically. There is commonly cross-tolerance between drugs of similar structure.
Failure of certain individuals to respond to normal doses of a drug, e.g. resistance to warfarin, vitamin D, constitutes a form of natural tolerance (see Pharmacogenetics, p. 101).
Pharmacokinetics
Pharmacokinetics3 is concerned with the rate at which drug molecules cross cell membranes to enter the body, to distribute within it and to leave the body, as well as with the structural changes (metabolism) to which they are subject within it.
The discussion covers the following topics:
• Drug passage across cell membranes.
• Order of reaction or process (first and zero order).
• Time course of drug concentration and effect:


• The individual processes: absorption, distribution, metabolism (biotransformation), elimination.
Drug passage across cell membranes
Passive diffusion
This is the most important means by which a drug enters the tissues and distributes through them. It refers simply to the natural tendency of any substance to move passively from an area of high concentration to one of low concentration. In the context of an individual cell, the drug moves at a rate proportional to the concentration difference across the cell membrane, i.e. it shows first-order kinetics (see p. 81); cellular energy is not required, which means that the process does not become saturated and is not inhibited by other substances.
It is useful to classify drugs in a physicochemical sense into:
• Those that are variably ionised according to environmental pH (electrolytes) (lipid soluble or water soluble).
• Those that are incapable of becoming ionised whatever the environmental pH (un-ionised, non-polar substances) (lipid soluble).
• Those that are permanently ionised whatever the environmental pH (ionised, polar substances) (water soluble).
Drugs that ionise according to environmental pH
• Acidic groups become less ionised in an acidic environment.
• Basic groups become less ionised in a basic (alkaline) environment and vice versa.
This in turn influences diffusibility because:
Permanently ionised drugs
The following are particular examples of the relevance of drug passage across membranes.
Maternal blood bathes the chorionic villi, which consist of a layer of trophoblastic cells that enclose fetal capillaries. Their large surface area and the high placental blood flow (500 mL/min) are essential for gas exchange, uptake of nutrients and elimination of waste products. Thus a lipid barrier separates the fetal and maternal bloodstreams, allowing the passage of lipid-soluble substances but excluding water-soluble compounds, especially those with a molecular weight exceeding 600.4
This exclusion is of particular importance with short-term use, e.g. tubocurarine (mol. wt. 772) (lipid insoluble) or gallamine (mol. wt. 891) used as a muscle relaxant during caesarean section do not affect the infant; with prolonged use, however, all compounds will eventually enter the fetus to some extent (see Index).
Carrier-mediated transport
Some carrier-mediated transport processes operate passively, i.e. do not require cellular energy, and this is facilitated diffusion, e.g. vitamin B12 absorption. Other, energy-requiring processes move substrates into or out of cells against a concentration gradient very effectively, i.e. by active transport; they are subject to saturation, inhibition and induction (see p. 91).
The order of reaction or process
• First-order processes by which a constant fraction of drug is transported/metabolised in unit time.
• Zero-order processes by which a constant amount of drug is transported/metabolised in unit time.
Zero-order processes (saturation kinetics)
As the amount of drug in the body rises, metabolic reactions or processes that have limited capacity become saturated. In other words, the rate of the process reaches a maximum amount at which it stays constant, e.g. due to limited activity of an enzyme, and any further increase in rate is impossible despite an increase in the dose of drug. In these circumstances, the rate of reaction is no longer proportional to dose, and exhibits rate-limited or dose-dependent5 or zero-order or saturation kinetics. In practice, enzyme-mediated metabolic reactions are the most likely to show rate limitation because the amount of enzyme present is finite and can become saturated. Passive diffusion does not become saturated. There are some important consequences of zero-order kinetics.
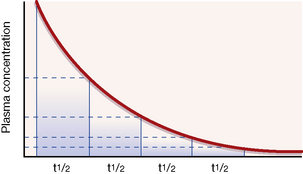
An illustration. Consider a man of average size who drinks about half (375 mL) a standard bottle of whisky (40% alcohol), i.e. 150 mL alcohol, over a short period, absorbs it and goes drunk to bed at midnight with a blood alcohol concentration of about 250 mg/dL. If alcohol metabolism were subject to first-order kinetics, with a t½ of 1 h throughout the whole range of social consumption, the subject would halve his blood alcohol concentration each hour (see Fig. 8.2). It is easy to calculate that, when he drives his car to work at 08.00 hours the next morning, he has a negligible blood alcohol concentration (less than 1 mg/dL) though, no doubt, a hangover might reduce his driving skill.
Time course of drug concentration and effect
Plasma half-life and steady-state concentration
Decrease in plasma concentration after an intravenous bolus injection
Following an intravenous bolus injection (a single dose injected in a period of seconds as distinct from a continuous infusion), plasma concentration rises quickly as drug enters the blood to reach a peak. There is then a sharp drop as the drug distributes round the body (distribution phase), followed by a steady decline as drug is removed from the blood by the liver or kidneys (elimination phase). If the elimination processes are first order, the time taken for any concentration point in the elimination phase to fall to half its value (the t½) is always the same; see Figure 8.2. Note that the drug is virtually eliminated from the plasma in five t½ periods.
Increase in plasma concentration with constant dosing
With a constant rate infusion, the amount of drug in the body and with it the plasma concentration rise until a state is reached at which the rate of administration to the body is exactly equal to the rate of elimination from it: this is called the steady state. The plasma concentration is then on a plateau, and the drug effect is stable. Figure 8.3 depicts the smooth changes in plasma concentration that result from a constant intravenous infusion. Clearly, giving a drug by regularly spaced oral or intravenous doses will result in plasma concentrations that fluctuate between peaks and troughs, but in time all of the peaks will be of equal height and all of the troughs will be of equal depth; this is also called a steady-state concentration, as the mean concentration is constant.6
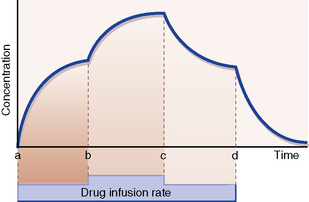
Time to reach steady state
of the ultimate steady state.
Change in plasma concentration with change or cessation of dosing
The same principle holds for change from any steady-state plasma concentration to a new steady state brought about by increase or decrease in the rate of drug administration. Provided the kinetics remain first order, increasing or decreasing the rate of drug administration (b and c in Fig. 8.3) gives rise to a new steady-state concentration in a time equal to 5 × t½ periods.
Similarly, starting at any steady-state plasma concentration (100%), discontinuing the dose (d in Fig. 8.3) will cause the plasma concentration to fall to virtually zero in 5 × t½ periods, as described in Figure 8.2.
Some t½ values appear in Table 8.1 to illustrate their range and implications for dosing in clinical practice.
Table 8.1 Plasma t½ of some drugs
| Drug | t½ |
|---|---|
| Adenosine | < 2 s |
| Dobutamine | 2 min |
| Benzylpenicillin | 30 min |
| Amoxicillin | 1 h |
| Paracetamol | 2 h |
| Midazolam | 3 h |
| Tolbutamide | 6 h |
| Atenolol | 7 h |
| Dosulepin | 25 h |
| Diazepam | 40 h |
| Piroxicam | 45 h |
| Ethosuximide | 54 h |
Therapeutic drug monitoring
Plasma concentration may correlate well with effect
Plasma concentration monitoring has proved useful:
• As a guide to the effectiveness of therapy, e.g. plasma gentamicin and other antimicrobials against sensitive bacteria, plasma theophylline for asthma, plasma ciclosporin to avoid transplant rejection, lithium for mood disorder.
• To reduce the risk of adverse drug effects when therapeutic doses are close to toxic doses (low therapeutic index), e.g. otic damage with aminoglycoside antibiotics; adverse CNS effects of lithium, nephrotoxicity with ciclosporin.
• When the desired effect is suppression of infrequent sporadic events such as epileptic seizures or episodes of cardiac arrhythmia.
• To check patient compliance on a drug regimen, when there is failure of therapeutic effect at a known effective dose, e.g. antiepilepsy drugs.
• To diagnose and manage drug overdose.
• When lack of therapeutic effect and toxicity may be difficult to distinguish. Digoxin is both a treatment for, and sometimes the cause of, cardiac supraventricular tachycardia; a plasma digoxin measurement will help to distinguish whether an arrhythmia is due to too little or too much digoxin.
Interpreting plasma concentration measurements
• The target therapeutic concentration range for a drug is a guide to optimise dosing together with other clinical indicators of progress.
• Take account of the time needed to reach steady-state dosing conditions (see above). Additionally, some drugs alter their own rates of metabolism by enzyme induction, e.g. carbamazepine and phenytoin, and it is best to allow 2–4 weeks between change in dose and meaningful plasma concentration measurement.
• As a general rule, when a drug has a short t½ it is desirable to know both peak (15 min after an intravenous dose) and trough (just before the next dose) concentrations to provide efficacy without toxicity, as with gentamicin (t½ 2.5 h). For a drug with a long t½, it is usually best to sample just before a dose is due; effective immunosuppression with ciclosporin (t½ 27 h) is obtained with trough concentrations of 50–200 micrograms/L when the drug is given by mouth.
Individual pharmacokinetic processes
Drug absorption into, distribution around, metabolism by and elimination from the body are reviewed.
Absorption
• Enteral: by mouth (swallowed) or by sublingual or buccal absorption; by rectum.
• Parenteral: by intravenous injection or infusion, intramuscular injection, subcutaneous injection or infusion, inhalation, topical application for local (skin, eye, lung) or for systemic (transdermal) effect.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


