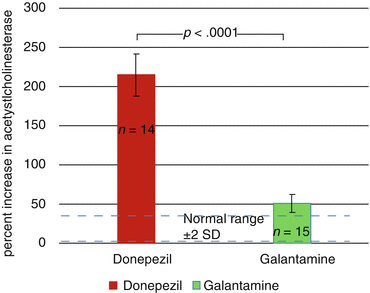
(1)
New York, New York, USA
3.1 Introduction
Despite numerous attempts to develop new classes of compounds for either the progression or symptomatic treatment of Alzheimer’s disease, there have been no successes to date. Hence, the mainstay of the treatment of mild-to-moderate Alzheimer’s disease (AD) remains the cholinesterase inhibitors. Despite the widespread use of these drugs, there is scant literature discussing the relative differences among these compounds and the practical consequences of those differences. Indeed, a recent review in a respected journal notes that “AD … responds only marginally and briefly to currently available drugs” (Bloom 2014). The purpose of this review article is to delineate the important properties that distinguish these compounds and the clinical implications of those differences. It will do so by largely focusing on donepezil, the most frequently prescribed cholinesterase inhibitor, and contrasting donepezil with galantamine, the cholinesterase inhibitor that differs the most in its mechanism of action within this class of compounds.
Two key properties differentiate donepezil and galantamine. These properties are the drugs’ interaction with nicotinic receptors and their half-lives. Galantamine has been shown to be a positive allosteric modulator of nicotinic receptors, a property not shared by donepezil (Samochocki et al. 2003). Galantamine has a half-life of 7–8 h (Product Monograph 2008). In contrast, donepezil’s half-life in the elderly is approximately 104 h (Ohnishi et al. 1993). The consequences of continuous, as compared to physiologically timed cholinesterase inhibition, will be addressed below.
3.2 Nicotinic Enhancement
Galantamine’s enhancement of nicotinic receptors is especially pronounced at the α4β2α5 receptor (Kuryatov et al. 2008). Galantamine potentiates depolarization of α4β2 nicotinic receptors in human embryonic kidney-263 cells (Samochocki et al. 2003). That effect is blocked by FK-1, an antibody that specifically binds the galantamine positive allosteric modulatory site on nicotinic receptors. Galantamine has a similar effect on α7 receptors in xenopus oocytes (Texido et al. 2005). Donepezil does not enhance the activity of nicotinic receptors beyond the effect of acetylcholinesterase inhibition. This difference suggests that galantamine should have a profile with more nicotinic activity than does donepezil. Conversely, donepezil should have a profile that favors more muscarinic activity than does galantamine. These differences, in nicotinic stimulation, and those in duration of action, to be discussed later, have profound clinical implications that are just being recognized.
3.2.1 Peripheral Cholinergic Effects
One simple way to differentiate nicotinic and muscarinic clinical effects is the relative incidence of diarrhea, as diarrhea is a reflection of muscarinic activity. The large, double-blind, placebo-controlled registration studies reported in the Physician’s Desk Reference indicate that the relative difference in the incidence of diarrhea between patients on rivastigmine or donepezil compared to placebo is large. Donepezil patients reported diarrhea 100 % more frequently than placebo patients (Medical Economics Staff 2002). In contrast, galantamine patients had only 29 % more diarrhea than their placebo counterparts. Table 3.1 summarizes these differences.
Table 3.1
Incidence of diarrhea in controlled clinical trials of cholinesterase inhibitors cited in 2002 Physicians’ Desk Reference (PDR)
Drug (dose) | PDR page # | Total n | % with diarrhea | % drug–% placebo (%) | ||
|---|---|---|---|---|---|---|
Drug | Placebo | Drug (%) | Placebo (%) | |||
Tacrine (40–160 mg) | 1354 | 634 | 342 | 16 | 5 | 11 |
Galantamine (16–24 mg) | 1796 | 1040 | 801 | 9 | 7 | 2 |
Rivastigmine (6–12 mg) | 2344 | 1189 | 868 | 19 | 11 | 8 |
Donepezil (5–10 mg)a | 2666 | 747 | 355 | 10 | 5 | 5 |
3.2.2 Cognitive Profile
The cognitive profiles of galantamine and donepezil demonstrate a difference that can be attributed to enhanced nicotinic stimulation by galantamine. A 52-week, rater-blinded study directly compared the effects of donepezil and galantamine on the Mini-Mental State Exam (MMSE) and the Alzheimer’s Disease Assessment Scale—cognitive (ADAS-cog) (Wilcock et al. 2003). In a preplanned analysis of patients meeting the UK’s National Institute of Clinical Excellence criteria for moderate AD (having MMSE scores from 10 to 18), the two drugs significantly differed in their ability to improve performance on the attention and language subscales over the 52 weeks of the study, as shown in Fig. 3.1. The language subscale contains commands, which require working memory. Both attention and working memory can be enhanced by nicotinic stimulation. On the ADAS-cog, performance on the “commands” question was also significantly better in galantamine than donepezil patients (data on file). This pattern of results with galantamine is consistent with its ability to enhance central nicotinic activity beyond cholinesterase inhibition, through allosteric modulation of receptors.
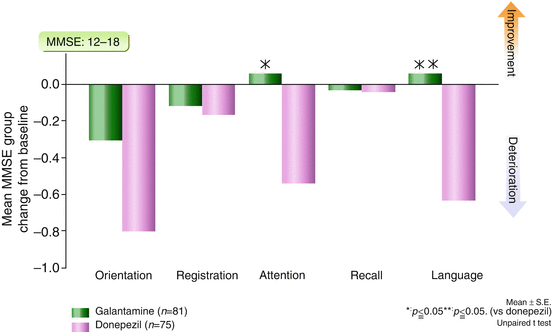
Fig. 3.1
Galantamine patients scored significantly better than donepezil patients on MMSE (Mini-Mental State Exam) subscales requiring attention and working memory, both of which involve nicotinic mechanisms. The drugs were compared in a randomized, 1-year, rater-blinded study in patients with MMSE scores of 12–18. MMSE Mini-Mental State Exam (Wilcock et al. 2003)
3.2.3 Neuroprotection
As the experimental therapeutics of AD has evolved, increased emphasis has been placed on the development of compounds that would offer neuroprotection, enhance the clearance of β-amyloid (Aβ), or decrease the formation or toxicity of various forms of the Aβ peptides. Nicotinic activity can mediate both neuronal survival and Aβ clearance. That nicotinic mechanisms may be neuroprotective has strong epidemiologic support in a preventive effect of smoking on the incidence of Parkinson’s disease. There is a beneficial effect of duration of smoking, and neuroprotection has been demonstrated in identical twins discordant for smoking (Chen et al. 2010). While lifestyle and many other physiological effects of smoking may contribute to these findings, the α4β2 and a7 nicotinic receptors can mediate neuroprotective mechanisms (Kawamata and Shimohama 2011). Smoking itself does not lower the incidence of AD, perhaps due to adverse effects on other organ systems (Kulkull 2001).
Depending on brain region, 11–37 % of α4β2 nicotinic receptors have as their fifth member, the α5 subunit (Kuryatov et al. 2008). These α4β2α5 subtypes are more sensitive to agonists and produce a larger maximal current than α4β2 receptors lacking the α5 subunit (Mao et al. 2008). Galantamine, at clinical concentrations, enhances the activity of α4β2α5 receptors by 220 %, as compared to 20–30 % for other nicotinic receptors (Kuryatov et al. 2008). As shown in Fig. 3.2, galantamine, applied simultaneously with a combination of glutamate and Aβ species, blocks their neurotoxic effect (Kihara et al. 2004). In a separate experiment, 24-hour pretreatment of a neuronal culture with galantamine increased survival following a toxic dose of glutamate by 78 % (p < .01). The protection against glutamate was nicotinic, as mecamylamine blocked 2/3 of galantamine’s benefit. Dihydrobetaerythroidine, an α4β2-blocker, reduced the galantamine effect by a little more than half, while methyllyaconitine, an α7-antagonist, caused a 1/3 reduction (all p < .01) (Takada-Takatori et al. 2006). Thus, galantamine’s potent effect on an especially active subset of α4β2 receptors may play a large part in galantamine-induced neuroprotection. Cholinesterase inhibitors which do not enhance nicotinic receptor function through allosteric enhancement may nevertheless be expected to have some nicotinic activity. Thus, donepezil can protect neurons against glutamate toxicity, but is 10× less potent than galantamine (Takada-Takatori et al. 2009).
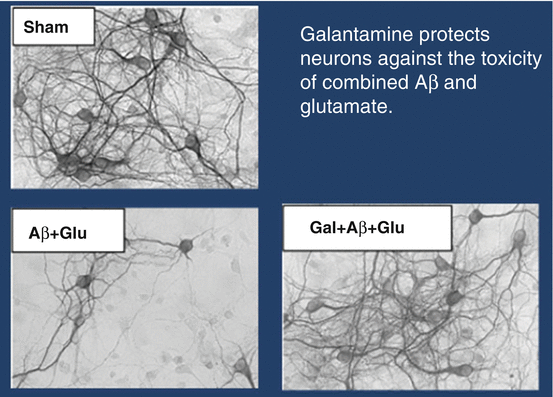
Fig. 3.2
Galantamine exerts a protective effect against β-amyloid (Aβ)-enhanced glutamate cytotoxicity. Aβ, Aβ1–40 (10.0 nM) + Aβ1–42 (1.0 nM) 4 days; Glu, glutamate (20.0 μM) 24 h; Aβ + Glu, 4-day treatment with Aβ followed by 24-h treatment with glutamate; Gal, simultaneous treatment with galantamine and Aβ for 4 days (1.0 and 10.0 μM). Galantamine 1.0 and 10.0 μM significantly protected neurons against Aβ-enhanced glutamate neurotoxicity (p < .01) (Kihara et al. 2004)
3.2.4 Aβ Clearance
Nicotinic stimulation can also enhance Aβ clearance. This becomes particularly relevant in light of studies that indicate that the key abnormality in Alzheimer’s disease of late-onset (LOAD) is the inability to adequately clear Aβ. When Aβ clearance was measured following the infusion of labelled leucine and cerebrospinal fluid (CSF) was sampled hourly, patients with LOAD were 30 % less efficient at clearing Aβ than controls (Mawuenyega et al. 2010). This finding took on increasing importance following a large study of brains from LOAD patients and controls. Bayesian analysis of the gene expression that differentiated these two groups indicated that upregulation of genes in the immune/microglial module in LOAD patients best differentiated them from controls. Gene expression in the immune/microglial module was most highly correlated with neuropathology traits such as frontal and parietal atrophy and ventricular enlargement. The authors note that alleles of genes found in genome-wide association studies to increase the risk of LOAD, such as CD33 and TREM2, also fall within the immune/microglial network. To further explore this finding, the central gene in the network, TYROBP, was overexpressed in microglial cells. This resulted in downregulation of 99 % of functional genes within the microglia, such as those involved in RNA metabolism and cell-cycle mitosis (Zhang et al. 2013). Microglia perform many functions which can variously exacerbate or attenuate the Alzheimer process. Microglial function may be impaired in LOAD patients.
The importance of the elucidation of a group of genes that influences immune modulation and microglial activity that differentiates LOAD patients from controls was underscored in an editorial discussing an experiment in which CD33, a microglial-surface protein increased in LOAD, inhibited Aβ clearance (Gandy and Heppner 2013). The editorial pointed out that microglia can exist in an inflammatory, harmful state, or an amyloid-clearing, helpful state and that “microglia-targeted therapies must be finely targeted.” It noted that there were only two approved drugs that were known to increase the phagocytic activity of microglia, the PPARγ agonist pioglitazone and the mixed acetylcholinesterase inhibitor–nicotinic allosteric agonist galantamine (Takata et al. 2010). This property is illustrated in Fig. 3.3 which demonstrates galantamine’s ability to promote Aβ clearance by interacting with its allosteric nicotinic modulatory site on microglia. The enhancement of Aβ clearance can be completely blocked by the antibody FK-1 which blocks the galantamine modulatory site on nicotinic receptors.
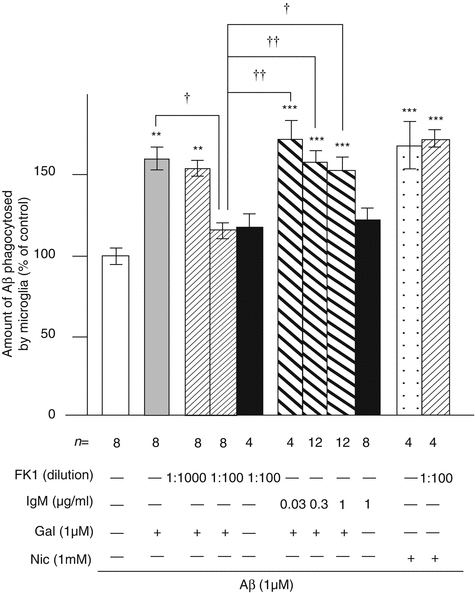
Fig. 3.3
Involvement of the APL-binding site for nAChRs in galantamine-enhanced microglial Aβ phagocytosis. Rat microglia were treated with 1 μM Aβ42 in the presence or absence of 1 μM galantamine or 1 mM nicotine. FK1 antibody or mouse IgM isotype control was added 10 min before treatment with Aβ42. The amounts of Aβ phagocytosed by microglia were measured by ELISA. **, p < .01; ***p < .001; versus Aβ42 alone. †, p < 0.05; ††, p < 0.01 versus Aβ42 plus galantamine and FK1 antibody (1:100). FK1 FK1 antibody, IgM mouse IgM isotype control, Gal galantamine, Nic nicotine, n number of samples (Takata et al. 2010)
Amyloid deposits in the brains of APdE9 mice carrying amyloid precursor protein (APP) and presenilin 1 (PS1) mutations, control and galantamine treated, are shown in Fig. 3.4. The brain slice shown in the left panel was from a mouse treated with galantamine, 5 mg/kg/day for two months prior to sacrifice at 11 months, resulting in a significant reduction in amyloid deposits. Additionally, treated mice showed improved learning and spatial memory in the water maze test (Takata et al. 2010). A similar result has been reported for short-term donepezil treatment of APP/PS1 mice (Easton et al. 2013). A 10-day treatment of Tg2576 (APPswe) mice with galantamine increased synaptophysin levels, but did not reduce Aβ species (Unger et al. 2006). These data indicate that cholinesterase inhibitors may be able to influence amyloid deposition in animal models of familial Alzheimer’s disease.
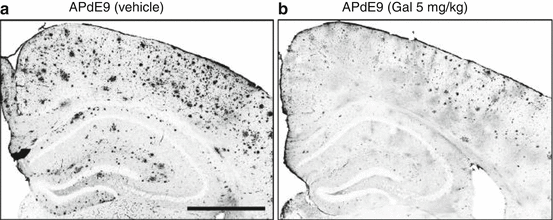
Fig. 3.4
Galantamine increased Aβ clearance in the brains of APdE9 mice. A and B, brain sections of vehicle-treated (a) or galantamine-treated (b) APdE9 mice were immunostained with anti-Aβ antibody. Mice were treated with 5 mg/kg daily of galantamine, or vehicle, for 2 months and sacrificed at 11 months. Scale bar, 500 μm (Takata et al. 2010)
3.3 Human Clinical Data
3.3.1 Biomarkers
Changes in amyloid dynamics under the influence of galantamine have been shown in humans as well. CSF Aβ was measured in a 3-month head-to-head study of galantamine, donepezil, and rivastigmine in mild-to-moderate AD patients (Nordberg et al. 2009). Patients were randomized to each of the three drugs, and CSF was collected at baseline and endpoint and analyzed by personnel blinded to treatment and sample order. CSF Aβ1–42 rose 17 % in galantamine patients, which was significantly different from the outcome in rivastigmine patients (Fig. 3.5). Figure 3.6 shows the CSF biomarker changes following treatment with the cholinesterase inhibitors in the context of typical healthy control and Alzheimer values from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database. The percentage changes in baseline CSF Aβ1–42 and phosphotau (ptau) which were reported by Nordberg et al. were applied to the mean CSF Aβ1–42 and ptau values for AD patients from the ADNI database (Okonkwo et al. 2010). The statistically significant differences between rivastigmine and galantamine in CSF Aβ1–42 and ptau are easily appreciated in Fig. 3.6, with rivastigmine moving biomarker values away from and galantamine moving Aβ1–42 and ptau towards healthy control values. Numerous compounds have cleared amyloid from various mouse animal models and have not altered the progression of the Alzheimer process. Biomarkers of Alzheimer’s disease still need further validation. The litmus test for whether any of these results has practical significance rests on clinical studies either in patients with AD or mild cognitive impairment (MCI). Since the average AD patient lives 8 years from the time of diagnosis, the most useful clinical studies are not the 6-month trials that have been used for registration purposes, rather they are trials of several years’ duration. Three placebo-controlled, randomized studies have been carried out in patients with MCI that report the effect of acetylcholinesterase inhibitors on the MRI biomarker, global brain atrophy. Over the course of 29 months, global atrophy was not significantly diminished in patients receiving donepezil compared to controls, nor was it at any time point in a four-year rivastigmine study (Jack et al. 2008; Feldman et al. 2007). In contrast, over 24 months MCI patients receiving galantamine had significantly less global atrophy than controls (Scheltens et al. 2004). It should be pointed out however that galantamine is not approved or recommended for patients with MCI (Winblad et al. 2008).
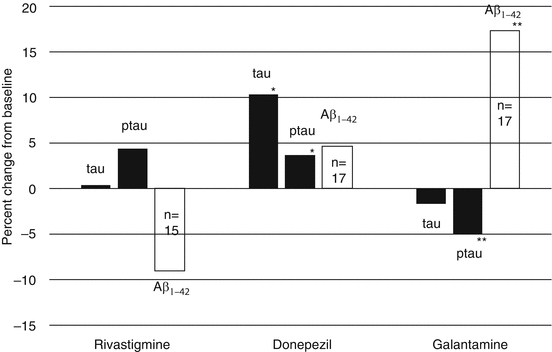
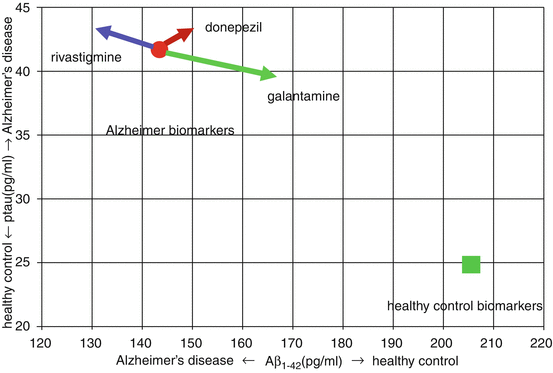
Fig. 3.5
Percent changes in CSF tau, ptau and Aβ1–42 in patients completing 13 weeks of treatment with galantamine, donepezil, or rivastigmine; *p < 0.05 versus baseline at 13 weeks, using one-way t-test; **p < 0.05 versus rivastigmine, using an ANOVA model with treatment as the factor and baseline value as the covariate. CSF cerebrospinal fluid, ptau phosphotau (Data from Nordberg et al. 2009)
Fig. 3.6
Changes in CSF Aβ1–42 and ptau following three months of rivastigmine, donepezil, or galantamine treatment of AD patients are shown in relation to average values for AD (circle) and healthy controls (square) from the ADNI database. Percentage changes from Nordberg et al. were applied to the ADNI baseline values. Significant differences in rivastigmine and galantamine effects on CSF Aβ1–42 and ptau are apparent, as rivastigmine moves biomarkers away from healthy control values, while galantamine moves biomarkers towards those of control subjects (Data from Nordberg et al. (2009) and Okonkwo et al. (2010))
3.3.2 Mortality
Thus, the data from the 2-year MCI trial of galantamine combined with the biomarker data support the notion that galantamine has properties that are not shared with other cholinesterase inhibitors, that this effect is likely mediated by the stimulation of allosteric nicotinic receptors, and that it could involve neuroprotection and/or amyloid clearance. However, by far the most compelling data differentiating galantamine from the other compounds in this class comes from a recently concluded two-year, placebo-controlled, randomized trial of galantamine in AD patients (Hager et al. 2014). This study entered 2045 patients with AD or AD with cerebrovascular disease. Thirty-five percent of these patients were male, their average age was 73, and their average MMSE score was 19. The demographic and baseline characteristics of the placebo and galantamine groups were similar.
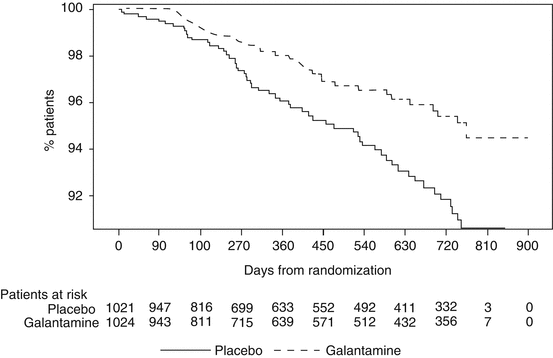
Before this study could be completed the Data Safety Monitoring Board halted the investigation because they had observed excess deaths in one arm of the study. Upon analysis it was determined that patients receiving placebo had a significantly higher mortality than patients receiving galantamine. Specifically, 56 deaths occurred in patients receiving placebo which was 5.5 % of that cohort. In contrast, there were 33 deaths in patients receiving galantamine, or 3.2 % of that cohort. The hazard ratio was .58, statistically significantly favoring galantamine (p = .01). The results are displayed in Fig. 3.7. The mortality benefit appears to increase with time.
Other large, double-blind, placebo-controlled studies of cholinesterase inhibitor administration to patients with mild-to-moderate Alzheimer’s dementia which have been conducted for varying periods have reported mortality. Those studies have been collapsed by duration of treatment and are presented in Table 3.2 (Rogers et al. 1998a, b; Burns et al. 1999; Tariot et al. 2000, 2001; Winblad et al. 2001; Mohs et al. 2001; AD 2000 Collaborative Group 2000; Raskind et al. 2000; Rockwood et al. 2001; Erkinjuntti et al. 2002; Brodaty et al. 2005; Homma et al. 2011; Hager et al. 2014). The data indicate that for a duration of drug administration of 6 months or less, there is a numerical diminution in mortality for patients taking either donepezil or galantamine compared to placebo. However, by 1 year, the advantage of donepezil on mortality is lost, and, by 2 years, the death rate in patients randomized to donepezil is 27–31 % higher than those randomized to placebo or to rivastigmine (Bullock et al. 2005). In contrast, the relative death rate on galantamine as compared to placebo at 6 months is maintained at 2 years.
Table 3.2
Mortality over time in double-blind, placebo-controlled, randomized trials in mild-to-moderate Alzheimer’s dementia
Donepezil | Galantamine | |||||
|---|---|---|---|---|---|---|
Donepezil deaths/n | Placebo deaths/n | Hazard ratio | Galantamine deaths/n | Placebo deaths/n | Hazard ratio | |
3–6 months | 7/1274 | 11/693 | 24/2803 | 16/1334 | ||
% | .55 % | 1.59 % | 0.35 | .86 % | 1.20 % | 0.71 |
1 year | 7/356 | 7/361 | ||||
% | 1.97 % | 1.94 % | 1.01 | |||
2 years | 63/242 | 50/244 | 33/1024 | 56/1021 | ||
% | 26.0 % | 20.5 % | 1.27 | 3.22 % | 5.49 % | 0.59 |
Mortality in severe and vascular dementias has been assessed in a number of 3–6-month studies. In three vascular dementia studies, 5–10 mg donepezil was administered to 1475 donepezil patients and 718 placebo patients. The mortality rates were 1.6 % for drug, and 1.1 % for placebo, a ratio of 1.46 (Black et al. 2003; Wilkinson et al. 2003; Roman et al. 2010). In contrast, the mortality ratio during a galantamine vascular dementia study was 0.49, as 5/396 (1.3 %) of galantamine patients and 10/390 (2.6 %) of placebo patients died (Auchus et al. 2007). In moderate-to-severe AD dementia studies, deaths totalled 28/773 (3.6 %) in donepezil patients and 32/669 (4.8 %) in placebo patients, a ratio of 0.76 (Black et al. 2007; Homma et al. 2008; Feldman et al. 2001; Winblad et al. 2006; Howard et al. 2007). Galantamine significantly reduced mortality in its one study in severe dementia. Death occurred in 8/207 (3.9 %) galantamine and 21/200 (10.5 %) placebo patients, a ratio of 0.37 (Burns et al. 2009). Thus, short-term mortality in galantamine-treated patients with dementias appeared to be favorably affected, which does not seem to be the case for donepezil use in vascular dementia.
In contrast to galantamine’s favorable results in dementia populations, a 2-year study of 16–24 mg galantamine and a 3-year study of 10 mg donepezil in MCI patients showed drug/placebo mortality ratios greater than unity. The mortality ratio for galantamine at 2 years was 1.7 (34/1026, 3.3 %, of galantamine, and 20/1022, 2.0 %, of placebo patients) (Winblad et al. 2008). The risk appeared to be nominally greatest earlier in the study, as shown in Figure 5 of Winblad et al. 2008. In the 3-year donepezil study, 7/259 (2.7 %) of donepezil, 5/253 (2.0 %) of placebo, and 5/257 (1.9 %) of vitamin E patients died, a donepezil/placebo ratio of 1.35, similar to that of the 2-year donepezil trial in AD patients (Petersen et al. 2005). These drugs are not recommended for use in MCI.
Returning to the substantial mortality reduction in patients with mild-to-moderate AD, a 42 % decrease with galantamine at 2 years needs an explanation. Serious adverse events occurring during or within 30 days of treatment did not differ between galantamine (12.6 %) and placebo patients (12.0 %). The hospitalization rate for galantamine patients, however, was 11 %, as compared to 8.6 % for placebo patients. The largest categories of serious adverse events and their hospitalization and death rates are presented in Table 3.3. It is apparent that there was no diagnostic category containing at least 2 % of the treatment-related serious adverse events whose mortality was relatively more diminished than any other. However, patients on galantamine who had serious treatment-emergent symptoms, except for neurological symptoms including AD, were hospitalized more frequently, and died less frequently, than placebo patients. This raises the possibility that galantamine patients responded differently from placebo patients when a serious adverse event occurred. The cognitive and functional effects of galantamine therapy are presented below.
Table 3.3
Serious treatment-emergent adverse eventsa ≥2 %, hospitalizations, and deaths
% incidence | % hospitalized | % death | ||||
|---|---|---|---|---|---|---|
pla | gal | pla | gal | pla | gal | |
Study totals | 12.0 | 12.6 | 8.6 | 11.0 | 4.6 | 3.0 |
Nervous system (e.g., AD, stroke) | 4.1 | 2.8 | 3.2 | 2.2 | 1.1 | 0.7 |
Cardiac (e.g., failure, myocardial infarction) | 2.4 | 2.2 | 0.8 | 1.2 | 1.8 | 1.3 |
Infections, infestations (e.g., pneumonia) | 1.5 | 2.0 | 0.6 | 1.7 | 0.5 | 0.4 |
Injury, poisoning, procedural complications | 2.2 | 2.0 | 1.8 | 2.0 | 0.2 | 0.1 |
3.3.3 Cognitive and Functional Outcomes
The results of the MMSE over the course of this study are presented in Fig. 3.8. The last observation carried forward, intent-to-treat analysis shows a highly statistically significant difference favoring galantamine over placebo at 6 and 24 months (p < .001). As measured by the Disability Assessment for Dementia (DAD), placebo patients took 17 months to reach the level of functional decline that galantamine patients experienced at 24 months. Mortality at 24 months in galantamine patients was at the level of placebo patients at 17 months as well. The MMSE and DAD subscales most affected by galantamine may offer insights into patients’ abilities and behaviors. The MMSE domains, in intention-to-treat analysis, which most differentiated galantamine from placebo patients were orientation, attention, and language. (Attention and language are the same scales noted above to differ between galantamine and donepezil patients and to utilize nicotinic mechanisms.) In the language question, a patient must remember and follow a 3-stage command, read and execute “Close your eyes,” and compose and write a sentence (Folstein et al. 1975). The DAD scales most significantly enhanced were basic and instrumental activities of daily living, initiation, effective performance, and planning and organization (data on file). The basic activities category includes eating, hygiene, and dressing, while telephoning, taking medications, and staying safely at home are instrumental activities (Gelinas and Gautier 1994). Initiation is the ability to decide or start an action appropriately. Effective performance is completing an action successfully. And planning and organization is essentially executive function—the ability to structure an activity, obtain supplies, make decisions, and solve problems during its execution. One can begin to appreciate that this subset of skills, which is maintained to the greatest degree with galantamine therapy, might help a person maintain health and obtain and cooperate with help when it is needed. Thus, galantamine’s ability to reduce cognitive and functional deterioration might have contributed to its mortality benefit.
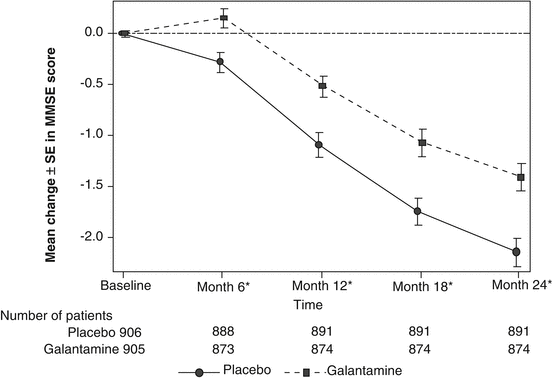
Fig. 3.8
Mean change in MMSE scores over time (LOCF) (ITT analysis set). *Significant difference between galantamine and placebo in MMSE score change from baseline. Estimates of treatment difference (95 % Cls) of MMSE using the repeated measures model (OC) were −0.48 (−0.73 to −0.22) at month 6, and −1.10 (−1.67 to −0.52) at month 24. Cl confidence interval, ITT intent-to-treat, LOCF last observation carried forward, MMSE Mini-Mental State Examination, OC observed case, SE standard error (Hager et al. 2014)
3.3.4 Galantamine and Memantine
Some insight into a possible biological mechanism underlying the clinical results with galantamine is revealed by the subpopulation of patients who was receiving concomitant treatment with memantine along with either galantamine or placebo. An analysis of the MMSE scores at month 24 broken down by the concomitant use or nonuse of memantine reveals a surprising result as indicated in Fig. 3.9. Memantine use completely blocked galantamine’s beneficial effect. It may simply be that memantine patients were sicker or unresponsive to cholinesterase inhibitors. Baseline MMSE values were significantly lower in memantine patients, by about a point. However, the decline of placebo-treated patients was similar whether or not memantine was taken. There is, however, a plausible pharmacological explanation. Memantine is an open-channel blocker of nicotinic receptors. Its distribution in the human brain is the same as that of the α4β2-selective compound 5-[125]-A-85380, being highest in the thalamus, followed by various cortical areas, with moderate binding in white matter (Ametamey et al. 2002; Pimlott et al. 2004). 18F-memantine distribution did not correspond to that of TCP, an uncompetitive NMDA receptor blocker. Thus, memantine binding followed a nicotinic but not a glutamate-receptor pattern. Memantine blocks α7 nicotinic receptors at an IC50 concentration of 5.1 μM and blocks α4β2 nicotinic receptors at an IC50 of 7 μM (Aracava et al. 2005; Buisson and Bertrand 1998). Memantine levels in plasma averaged 120 ng/ml during phase III studies in humans, about 0.67 μM (Periclou et al. 2006). Memantine partitions nearly 30× to brain tissue over plasma in rats (Saab and Roder 2011). Brain to plasma partitioning is similar in humans, as a 25:1 ratio was reached at the end of the PET study, at which time brain levels were still rising. All of these data taken together suggest that, in clinical use, memantine concentrations in brain tissue are well over 15 μM, blocking the nicotinic receptors whose function galantamine enhances. Thus, memantine negates galantamine’s positive effect on the MMSE. This result would be consistent with an important role for nicotinic mechanisms in the cognitive effects of galantamine. The basic science therefore suggests that, in the clinic, galantamine may not have an effect in the presence of memantine, and this may be an inadvisable combination.