Fundamental Concepts and Terminology
OBJECTIVES
The reader will be able to:
- Define the following terms: agonist, all-or-none response, antagonist, baseline, bioavailability, biomarker, clinical response, compartment, cumulative frequency, disease progression, disposition, distribution, elimination, endogenous, enterohepatic cycling, excretion, exogenous, extravascular administration, first-pass loss, fractional turnover rate, full agonist, full antagonist, graded response, intestinal absorption, intravascular administration, local administration, maximum effect, metabolic intercon-version, metabolism, metabolites, parenteral administration, partial agonist, partial antagonist, pharmacokinetic–pharmacodynamic (PK/PD) modeling, placebo, pool size, potency, prodrug, quantal response, safety biomarker, specificity, steepness factor, surrogate endpoint, systemic absorption, turnover, turnover rate, turnover time, up-or-down regulation.
- Discuss the limitations to interpretation of pharmacokinetic data imposed by assays that fail to distinguish between compounds administered (e.g., R- and S-isomers) or between drug and metabolite.
- Show the general contribution of mass balance concepts to drug absorption and drug and metabolite disposition.
- Explain why plasma drug concentration can serve as a useful correlate of response.
- Briefly discuss issues involved in the assessment of drug effect.
- Describe, with examples, how the intensity of a graded response changes with concentration at the site of action.
- Describe the parameters of the model that often characterize the relationship between a graded response and plasma concentration.
- Describe how a quantal response is related to plasma concentration.
- Discuss the relative value of potency, maximum effect, and specificity in drug therapy.
- Briefly discuss how the condition being treated influences the relative importance of the features of the exposure–time profile after a single oral dose.
 his chapter introduces input–exposure (pharmacokinetics) and exposure–response (pharmacodynamic) relationships and defines the terms commonly used in these areas. Concepts in these areas play many key roles, as listed in Table 2-1, in the development and use of medicinal agents. The chapter begins with a definition of systemic exposure and consideration of basic models and methods for evaluating it.
his chapter introduces input–exposure (pharmacokinetics) and exposure–response (pharmacodynamic) relationships and defines the terms commonly used in these areas. Concepts in these areas play many key roles, as listed in Table 2-1, in the development and use of medicinal agents. The chapter begins with a definition of systemic exposure and consideration of basic models and methods for evaluating it.

Before considering the subject of exposure, a distinction must be made between those drugs that act locally and those that act systemically. Locally acting drugs are administered at the local site where they are needed. Examples of products intended for local administration are eye drops, nasal sprays, intravaginal creams, and topical preparations for treating skin diseases. This chapter, and indeed much of the book, emphasizes those drugs that act within the blood or that must be delivered to the site of action by the circulatory system. In general terms, we say such drugs act systemically.
PHARMACOKINETICS
SYSTEMIC EXPOSURE
Needless to say, the response produced by a systemically acting drug is related, in one way or another, to the amount entering the body and its duration there. While responses can be observed, often noninvasively, such as monitoring blood pressure, drug within the body, which causes the change in blood pressure in the first place, is usually monitored invasively. To understand the relationship between the administration of drugs and the responses produced, we need to follow the events occurring within the body with time. A key to doing so is to measure the body’s internal exposure to the drug. Here, our options are limited. It is rare when we can measure drug directly at the site of action, such as the brain or heart. Rather, we turn to more accessible sites to assess the systemic exposure to the drug.
Sites of Measurement. Various sites of exposure assessment are used, including plasma, serum, whole blood, breath, milk, saliva, and urine. The first three fluids (plasma, serum, and blood) are obtained by invasive techniques. The differences among plasma, serum, and whole blood drug concentrations are shown in Table 2-2. Breath, milk, saliva, and urine are obtained noninvasively. Drug concentrations at these sites reflect the concentrations in plasma, blood, or serum.
The two fluids most commonly sampled are blood and urine. Drug within blood generally equilibrates rapidly with drug in blood cells and on plasma proteins. Accordingly, any of the fluids (whole blood, plasma, or serum) can be used to reflect the systemic time course of drug. In practice, plasma is preferred over whole blood primarily because components in blood cause interference in many assay techniques. Plasma and serum usually yield equivalent drug concentrations, but plasma is considered easier to prepare, as blood must be allowed to clot to obtain serum. During this process, hemolysis can occur, producing a concentration that is neither that of drug in plasma or blood, or causing interference in the assay.
Unbound Drug Concentration. A comment is in order at this stage with respect to the binding of drugs to plasma proteins. The total plasma (or serum) concentration includes both unbound and bound drug. Drug distribution and elimination, as well as pharmaco-dynamic responses, are dependent on the unbound concentration. It is only the unbound drug that passes through cell membranes to reach sites of storage, metabolism, or activity. Because the ratio of unbound to total drug, fraction unbound, usually does not change, it makes little difference whether total or unbound drug is measured. However, in those conditions in which binding is altered, such as the presence of another drug that displaces the drug of interest from its binding protein, renal disease, hepatic disease, surgery, severe burns, and pregnancy, measurement of the unbound concentration becomes important.

Exposure–Time Profile. The systemic exposure–time profile is a function of the rate and extent of drug input, distribution, and elimination. Figure 2-1 shows the salient features of the systemic exposure–time profile after ingesting a single oral dose. The highest concentration is called the maximum concentration, Cmax, or maximum systemic exposure. The time of its occurrence is called the time of maximum concentration, tmax, or time of maximum exposure. Both Cmax and tmax depend on how quickly the drug enters into and is eliminated from the body. The area under the concentration-time curve over all time, abbreviated as AUC, is a measure of the total systemic exposure to the drug. The concentration profile before the peak is a function of how quickly the drug enters the systemic circulation; after the peak, it is a function of how quickly the drug is eliminated.
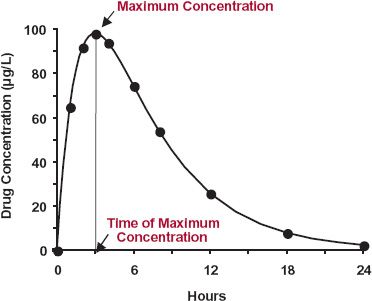
FIGURE 2-1. Drug concentration–time curve following a single oral dose showing the maximum systemic exposure (Cmax) and the time of its occurrence (tmax). The concentration could represent drug in whole blood, plasma, or serum.
The measured peak concentration (peak systemic exposure) of the example in Fig. 2-1 is 96 μg/L and the peak time is 3.0 hr. To get the AUC, the concentration must be integrated with respect to time. One common way to accomplish this is by approximating the areas between measurements as trapezoids and summing up the areas of the successive trapezoids (see Appendix A for further details).
Period of Observation. A basic kinetic principle is to make observations within the time frame of interest. An atomic physicist measures atomic events that occur within microseconds or nanoseconds. A geologist, on the other hand, may be studying plate tectonics, which occur on the scale of hundreds of thousands to millions of years. To appreciate the events, both of these individuals must make their observations consistent with the time frame of the kinetic events.
The same principles apply to drugs. Sometimes, drugs have a very short sojourn within the body. Following an intravenous (i.v.) bolus dose of such drugs, most of the dose may leave the body within minutes to hours. Other drugs remain in the body for days, weeks, months, and in a few instances, years. The period of measurement of systemic exposure must then be tailored to the kinetics of the specific drug of interest.
CHEMICAL PURITY AND ANALYTIC SPECIFICITY
A general statement needs to be made about the chemical purity of prescribed medicines and the specificity of chemical assays.
Over the years, a major thrust of the pharmaceutical industry has been to produce therapeutic agents and products that are not only as safe and effective as possible, but well characterized to ensure reproducible qualities. The majority of administered drug products today are therefore prepared with essentially pure drugs and, coupled with specific analytic techniques for their determination in biologic fluids, definitive information about their pharmacokinetics is gained. However, a large number of drug substances are not single chemical entities but rather mixtures. This particularly applies to stereoisomers and proteins. The most common stereoisomers found together in medicines are optical isomers, or compounds for which their structures are mirror images; the drug substance is often a racemate, a 50:50 mixture of the R- and S-isomers. Some drug substances contain geometric isomers (e.g., cis and trans) and still others, especially proteins of high molecular weight derived from natural products or through fermentation, may be a mixture of structurally related, but chemically distinct, compounds. Each chemical entity within the drug product can have a different pharmacologic, toxicologic, and/or pharmacokinetic profile. Sometimes these differences are small and inconsequential, other times the differences can be therapeutically important. For example, dextroamphetamine (S-isomer) is a potent central nervous stimulant, whereas the R-isomer is almost devoid of such activity. Sometimes the exposure–time profiles of the enantiomers are very different when given orally in racemic form, as shown in Fig. 2-2 for aceno-coumarol, an oral anticoagulant, and methylphenidate, used to treat hyperactivity in children (reasons for the differences are explained in the “Study Problems” of Chapter 6). Despite the occasional occurrence of such striking differences in pharmacokinetics or pharmacodynamics, historically, many chemical assays have not distinguished between stereoisomers. Obviously, under these circumstances, attempting to quantify the various processes and to relate plasma concentration to response has many problems with no simple solution. Notwithstanding these problems, specific information about each chemical entity should be sought whenever possible. To avoid these problems, stereospecific assays are now more commonly used to measure individual isomers, and stereoisomers are being developed and marketed as single chemical entities, such as S-naproxyn, a nonsteroidal anti-inflammatory agent, and tamsulosin (R-isomer: Flomax), an a-adrenergic blocking agent used to treat benign prostatic hyperplasia. In contrast, many new protein and polypeptide drugs are being introduced that may, in many instances, lack high purity. Furthermore, these latter substances are often measured by assays that lack chemical specificity.
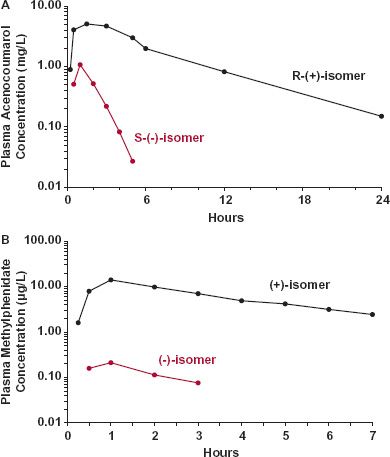
FIGURE 2-2. A. The exposure–time profiles of the R-(+) and S-(−) isomers of acenocoumarol, an agent used in some countries, differ strikingly after oral administration of 20 mg of a 50/50 racemic mixture to a subject. (From: Gill TS, Hopkins KJ, Rowland M. Stereospecific assay of acenocoumarol: application to pharmacokinetic studies in man. Br J Clin Pharmac 1988;25:591–598.) B. The peak and total systemic exposures to the (−) isomer of methylphenidate (colored) is negligible compared to the (+) isomer (black) following the oral administration of 30 mg of a racemic mixture of the two isomers, the common form of the drug. (From: Aoyama T, Kotaki H, Sasaki T, et al. Nonlinear kinetics of threo-methylphenidate enantiomers in a patient with narcolepsy and in healthy volunteers. Eur J Clin Pharmacol 1993;44:79–84.)
Some medicinal products contain several active compounds, an example being the combination of a thiazide diuretic and a β-blocker to lower blood pressure, each acting by a different mechanism. Specific assays are therefore needed to follow the pharmacokinetics of each compound. Herbal preparations contain a myriad of compounds, some of which may well be active. However, given the variable composition of such preparations and the uncertainty as to what might contribute to any observed effect, attempting to characterize the input–exposure relationships for many herbal products is highly problematic.
An added concern exists following drug administration, namely, the formation of metabolites, compounds formed from the drug usually by enzymatic reactions. To be of value, an analytic procedure must distinguish between drug and metabolite(s), many of which have very different pharmacokinetic and pharmacodynamic properties compared to the parent compound. Today, most assays have this desired specificity, except for many of those used to measure protein and polypeptide drugs.
A potential problem exists when using radiolabeled drugs to determine the fate of drug and related materials following drug administration. Incorporation of one or more radionuclides, usually 14C and 3H, into the molecular structure allows for simple and ready detection within a complex biologic milieu, but not necessarily of the administered drug. Complete recovery of all of a radiolabeled dose in urine, following oral drug administration, is useful in identifying the ultimate location of drug-related material but may provide little to no kinetic information about the drug itself. Consider, for example, a case in which the entire dose of an orally administered drug is destroyed in the gastrointestinal tract, yet degradation products enter the body and are ultimately excreted into the urine. Full recovery of radioactivity may suggest that the drug is completely available systemically when, in fact, none is. A basic lesson is learned here: Distinguish carefully between drug and metabolite(s). Each chemical entity must be considered separately for kinetic data to be meaningful.
ACTIVE METABOLITES AND PRODRUGS
Metabolites are sometimes thought of as weakly active or inactive end-products. For many drugs this is the case, but for many others it is not. Sometimes, the compound administered is an inactive prodrug that must be metabolized to the active compound. A prodrug is often developed intentionally to overcome an inherent problem with the active drug, such as poor or highly variable oral absorption. Examples of prodrugs include dolasetron mesylate (Anzemet), used to prevent chemotherapy-induced nausea and vomiting, famci-clovir (Famvir), an antiviral agent, and most of the angiotensin-converting enzyme inhibitors, used to lower blood pressure. Many drugs have metabolites that augment the activity of the administered compound, as is the case with sildenafil (Viagra), an agent used to treat erectile dysfunction. Some drugs form metabolites that produce other effects. For example, acyl glucuronides of a number of drugs (polar esters of the drug formed by its conjugation with glucuronic acid), including several nonsteroidal anti-inflammatory agents, react with some proteins and have been implicated in a wide range of adverse effects, including hypersensitivity reactions and cellular toxicity. Metabolites may also, by inhibiting drug metabolism, prolong or enhance drug response, or when acting to increase drug metabolism, shorten or diminish drug response. Delavirdine mesylate (Rescriptor), a reverse transcriptase inhibitor of the human immunodeficiency virus type 1 (HIV-1), is an example of the former.
Drug (or prodrug) and metabolites often have very different systemic exposure–time profiles, as shown in Fig. 2-3 for aspirin and its primary metabolite, salicylic acid. It is well to remember that therapeutic or adverse effects may reside with either the compound administered or with one or more of the metabolites. Differences in their exposure–time profiles can, therefore, have a major impact on drug response. This topic is expanded on in Chapter 20, Metabolites and Drug Response.
ANATOMIC AND PHYSIOLOGIC CONSIDERATIONS
As previously stated, measurement of a drug in the body is usually limited to plasma and occasionally, blood. Nonetheless, the information obtained at these sites has proven to be very useful. Such usefulness can be explained by anatomic and physiologic features that affect a drug’s sojourn within the body following its administration.
Blood is the most logical site for measurement of drug in the body. Blood receives drug from the site of administration and carries it to all the organs, including those in which the drug acts and those in which it is eliminated. This movement of drug is depicted schematically in Fig. 2-4 (page 24).
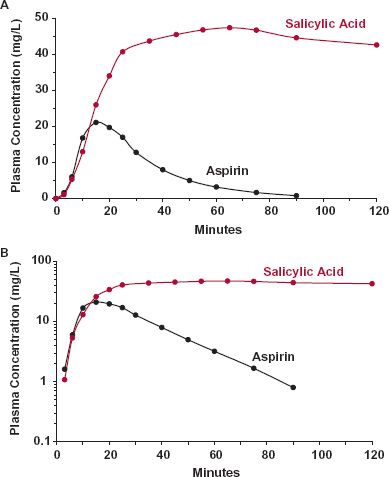
FIGURE 2-3. The exposure–time profile of a metabolite can differ greatly from that of the administered drug, as shown for salicylic acid (colored) and aspirin (black) on linear (A) and semilogarithmic (B) plots following the oral ingestion of 650 mg of aspirin. Consideration of the exposure–time profiles of metabolite(s) becomes particularly important therapeutically when the metabolite contributes to the therapeutic response or is associated with adverse events. When aspirin is given for its anticlot-ting activity, all the therapeutic activity lies with the drug, but when given for anti-inflammatory or antipyretic effects, most of the activity lies with the metabolite. (From: Rowland M, Riegelman S, Harris PA, Sholkoff SD. Absorption kinetics of aspirin in man following oral administration of an aqueous solution. J Pharm Sci 1972;61:379–385.)
Sites of Administration. There are several sites at which drugs are commonly administered. These sites may be classified as either intravascular or extravascular. Intravascular administration refers to the placement of a drug directly into the blood (i.e., either intravenously or intra-arterially).
Extravascular modes of administration include the buccal (between the cheek and the gums), intradermal, intramuscular, oral, pulmonary (inhalation), subcutaneous (into fat under the skin), rectal, and sublingual (under the tongue) routes, as listed in Table 2-3. Whether given through the alimentary canal (food canal or gastrointestinal tract) or any other extravascular route, an additional step, absorption, is required for drug to reach the systemic site of measurement relative to that required after intravascular administration.
Another term often used is parenteral administration. This refers to administration apart from the intestines. Today, the term is generally restricted to those routes of administration in which drug is injected through a needle. Thus, parenteral administration includes intramuscular, intravascular, and subcutaneous routes. Although the use of other routes, such as skin patches or nasal sprays (for systemic delivery), are strictly forms of parenteral administration, this term is not used for them.
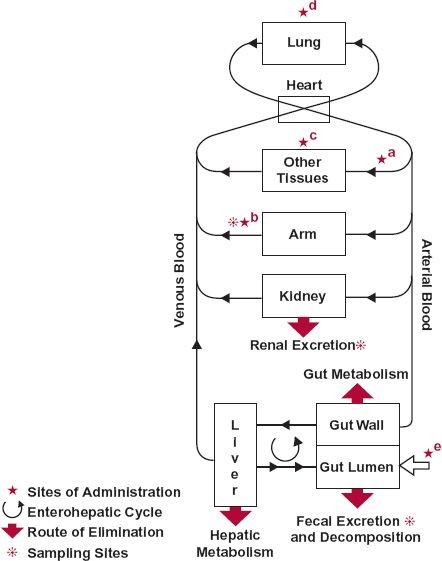
FIGURE 2-4. Once absorbed from any of the many sites of administration, drug is conveyed by blood to all sites within the body including the eliminating organs and site(s) of action. Sites of administration include: a, artery; b, peripheral vein; c, muscle and subcutaneous tissue; d, lung; and e, gastrointestinal tract, the open arrow. When given intravenously into an arm vein, the contralateral arm should then be used for sampling. The movement of virtually any drug can be traced from site of administration to site(s) of elimination.

Drug may also be administered regionally (e.g., into the pleural or peritoneal cavities or into the cerebrospinal fluid). Regional administration also includes intra-arterial injection into the vessel leading to a tissue to be treated (e.g., a cancerous tumor). It is a potential means of gaining a selective therapeutic advantage. This advantage, in comparison with other routes of administration, comes about by increasing drug exposure locally, where it is needed, and decreasing or producing little or no change in exposure throughout the rest of the body, where it is not wanted.
Events After Entering Systemically. Once entered systemically, a drug is distributed to the various organs of the body by the blood. Distribution is influenced by organ blood flow, organ size, binding of drug within blood and in tissues, and transport across tissue membranes.
The two principal organs of elimination, the liver and the kidneys, are shown separately in Fig. 2-4. The kidneys are the primary organs for excretion of the chemically unaltered or unchanged drug. The liver is the usual organ for drug metabolism; however, the kidneys, the intestinal tissues, and other organs can also play an important metabolic role for certain drugs. The metabolites so formed are either further metabolized or excreted unchanged, or both. The liver may also secrete unchanged drug into the bile. The lungs are, or may be, an important route for eliminating volatile substances, for example, gaseous anesthetics. Another potential route of elimination (not shown) is lactation. Although generally an insignificant route of elimination in the mother, drug in the milk may be consumed in sufficient quantity to affect a suckling infant.
Drugs are rarely given alone as the pure substance. They are formulated into a product that is convenient for the patient and optimizes the drug’s performance when administered. For example, tablets and capsules permit a fixed dose to be easily administered orally. The formulation of the product may result in rapid release of the active drug, or its release may be modified to diminish fluctuation in systemic exposure between doses or to allow less frequent dosing.
MODELS FOR DRUG ABSORPTION AND DISPOSITION
A model is useful to summarize data and facilitate extrapolation and prediction after drug administration. The complexities of human anatomy and physiology would appear to make it difficult, if not impossible, to model how the body handles a drug. Perhaps surprisingly then, it is a simple pharmacokinetic model, depicted in Fig. 2-5, which has proved useful in many applications and is emphasized throughout much of this book.
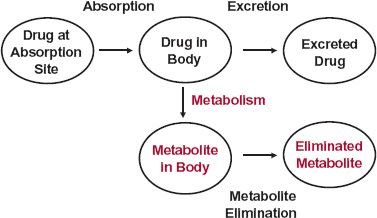
FIGURE 2-5. A drug is simultaneously absorbed into the body and eliminated from it, by excretion and metabolism. The processes of absorption, excretion, and metabolism are indicated with arrows and the compartments with ovals. The compartments represent different locations and different chemical species (color = metabolite). Metabolite elimination may occur by further metabolism or excretion.
The boxes in Fig. 2-5 represent compartments that logically fall into two classes: transfer and chemical. The site of administration, the body, and excreta are clearly different places. Each place may be referred to as a location or transfer compartment. In contrast, metabolism involves a chemical conversion; drug and metabolite in the body are clearly in compartments that differ chemically, but not by location. The same principle applies to excreted drug and metabolite.
The model is based on amounts of drug and metabolite. However, they can only be directly measured in urine and feces. The total amount of drug metabolized includes metabolites in, as well as eliminated from, the body. The amount in the body is usually determined from measurement of the blood or plasma concentration. Estimates of drug in the absorption compartment are also usually made indirectly from either blood or urine data. Drug at the absorption site includes that which is never absorbed, for example, drug that is ultimately decomposed in the gastrointestinal tract or lost in the feces after oral administration.
The model is readily visualized from mass balance considerations. The dose is accounted for at any one time by the molar amount of substance in each of the compartments:
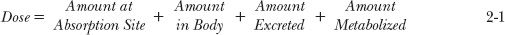
The mass balance of drug and related material with time is shown in Fig. 2-6. Because the sum of the molar amounts of drug in transfer and chemical compartments is equal to the dose, the sum of the rates of change of the drug in these compartments must be equal to zero so that:
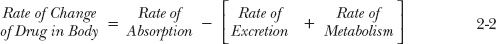
The relationships expressed in Eqs. 2-1 and 2-2 apply under all circumstances, regardless of the nature of the absorption and elimination processes. They are particularly useful in developing more complex models for quantifying drug absorption and disposition. Pharmacokinetics is the quantitation of the time course of a drug and its metabolites in the body and the development of appropriate models to describe observations and predict outcomes in other situations.
Examples of other common models used in pharmacokinetics are shown in Fig. 2-7. One form (A) is the use of equations, which may consist of either integrated or differential equations, or both. Physiologic models (B) are also common, such as those depicting movement of a drug through a tissue or organ of elimination. Figure 2-4 is such a physiologic model of drug absorption and disposition in the whole body. The disposition of a drug is often simulated using compartmental models (C) for drug distribution within the body. Shown are one- and two-compartment models. The two-compartment model, in which drug distributes from a central compartment, of which the plasma is a part, to a peripheral compartment, which is generally viewed as the tissues, is a very common one.
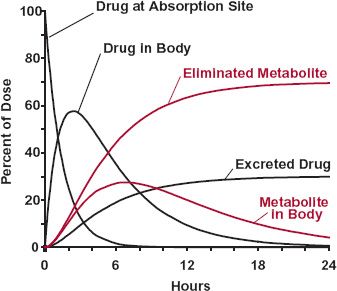
FIGURE 2-6. Time course of drug and metabolite in each of the compartments shown in Fig. 2-5. The amount in each compartment is expressed as a percentage of the dose administered. In this example, all the dose is absorbed. At any time, the sum of the molar amounts in the five compartments equals the dose.
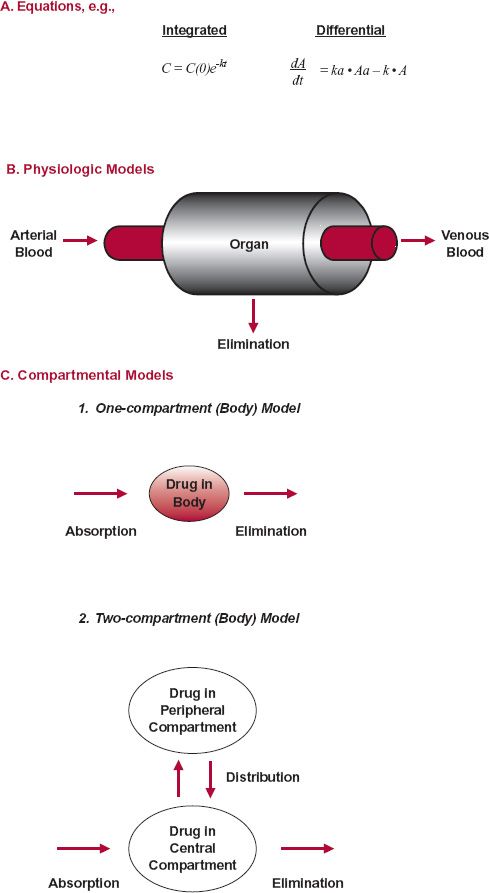
FIGURE 2-7. Several kinds of models are used in pharmacokinetics. A. Differential and integral eqs. can be one way of modeling the kinetic behavior of drugs in the body. B.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


