Follicular Carcinoma
Yuri E. Nikiforov
N. Paul Ohori
DEFINITION
Follicular carcinoma is a malignant well-differentiated tumor of thyroid follicular cells that lacks the diagnostic nuclear features of papillary carcinoma. When used without additional qualifiers, this term typically refers to the conventional type of follicular carcinoma. Oncocytic (Hürthle cell) follicular carcinoma represents the main histopathologic variant of follicular carcinoma.
INCIDENCE AND EPIDEMIOLOGY
Follicular carcinoma is the second most common type of thyroid cancer after papillary carcinoma. The data collected over the past 20 to 30 years indicate that follicular carcinoma, when combining both the conventional type and oncocytic variant, accounts for approximately 15% of all thyroid cancer cases.1 In recent years, this number has decreased to 10% and even lower.1,2 Several reasons are likely to be responsible for this trend. First, the incidence of thyroid papillary cancer has been on a rise in many countries around the world; it has practically tripled in the United States over the past 30 years, making the frequency of follicular carcinoma proportionally smaller.1 Second, as described in the next section, a higher incidence of follicular carcinoma is associated with dietary iodine deficiency. Severe iodine deficiency has been largely eliminated or diminished by widespread iodization of salt and other food supplies. Third, in the late 1980s to early 1990s, diagnostic histopathologic criteria for the follicular variant of papillary carcinoma became better recognized. As a result, many follicular-pattern carcinomas that would be previously diagnosed as follicular carcinomas are now classified as papillary carcinoma.2,3
The absolute incidence of follicular carcinoma has not changed significantly during the last several decades.1,4,5 On the basis of the Surveillance, Epidemiology and End Results (SEER) database, the annual incidence in the United States during the time interval between 1973 and 2008 remained at 0.8 to 1.1 per 100,000 persons per year.1
Most follicular carcinomas are tumors of conventional type, and 25% to 30% are of oncocytic type.1,6,7 Females are more commonly affected than males, with a gender ratio of approximately 2.5:1 for conventional and oncocytic types of follicular carcinomas based on the SEER database.1 Some studies have found a slightly lower female predominance among oncocytic carcinomas (1.7:1).7,8 These tumors are rare in children. Their incidence increases with age, reaching a peak during the fifth decade of life for conventional follicular carcinomas (Fig. 10.1). For oncocytic tumors, the incidence continues to climb beyond the fifth decade of life. In fact, more than half of oncocytic follicular carcinomas are diagnosed in patients older than 60 years.8
ETIOLOGIC FACTORS
Etiologic factors implicated in the development of this tumor are summarized in Table 10.1. Most follicular carcinomas show no familial occurrence and occur as sporadic tumors. Two environmental factors implicated in the etiology of this disease are iodine deficiency and exposure to ionizing radiation. Residing in areas of low-dietary iodine intake is associated with an up to 2- to 3-fold increase in risk when compared with areas of sufficient iodine consumption.9,10,11 Active iodine supplementation in the regions of severe iodine deficiency has resulted in a significant decrease of follicular carcinoma incidence, which provides additional
evidence for this etiologic factor.12,13 Low-iodine diet is associated with the increased thyroid-stimulating hormone (TSH) stimulation and development of thyroid hyperplasia in a form of endemic goiter, both of which are likely to be involved in promoting follicular carcinogenesis. A low-iodine diet administered to laboratory animals produces thyroid hyperplasia and nodular disease, followed by carcinoma development in some animals, simulating the disease progression in humans.14,15
evidence for this etiologic factor.12,13 Low-iodine diet is associated with the increased thyroid-stimulating hormone (TSH) stimulation and development of thyroid hyperplasia in a form of endemic goiter, both of which are likely to be involved in promoting follicular carcinogenesis. A low-iodine diet administered to laboratory animals produces thyroid hyperplasia and nodular disease, followed by carcinoma development in some animals, simulating the disease progression in humans.14,15
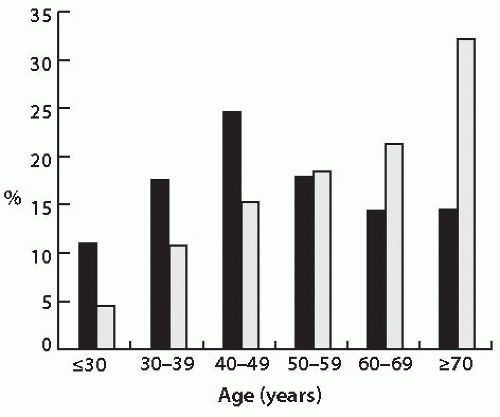 FIGURE 10.1. Age distribution of patients with conventional type follicular carcinoma (black bars) and oncocytic follicular carcinoma (gray bars). Based on the data reported by Hundahl SA, Cady B, Cunningham MP, et al. Initial results from a prospective cohort study of 5583 cases of thyroid carcinoma treated in the United States during 1996. U.S. and German Thyroid Cancer Study Group. An American College of Surgeons Commission on Cancer Patient Care Evaluation study. Cancer. 2000;89:202-217. |
Table 10.1 Etiologic Factors for Thyroid Follicular Carcinomas | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
Exposure to ionizing radiation is associated with predisposition to follicular carcinoma, although the risk is not as high as it is for papillary carcinoma. Cumulative analysis of several irradiated cohorts has demonstrated the relative risk of 5.2 for radiation-induced follicular carcinoma, about half the risk for papillary cancer.16 In the United States, a history of radiation exposure is found in approximately 4% of follicular carcinoma patients.8
Preexisting benign thyroid disease is a well-established risk factor for follicular carcinoma and is identified in approximately 15% of patients.8 Benign thyroid nodules/adenomas confer a higher risk as compared with nontoxic goiter (relative risk 60 to 70 vs. 7 to 8).17,18 The risk is highest during the first 2 years after diagnosis of a benign thyroid disease, and it persists for >10 years. Preexisting benign thyroid disease may affect the incidence of carcinomas in several ways. Follicular adenoma may serve as a direct precursor lesion for follicular carcinoma. This has been directly demonstrated in mice lacking the PTEN gene function,19 and further evidence for the follicular adenoma to follicular carcinoma progression is discussed later in the chapter. Goiter may predispose to follicular carcinoma by increasing the rate of cell proliferation owing to prolonged TSH stimulation, which enhances the chance for mutations in dividing cells.20 This is supported by more common occurrence of follicular carcinomas than papillary carcinomas in patients with dyshormonogenetic goiter.21,22 This condition is caused by an inherited defect in thyroid hormone production that leads to highly elevated TSH levels and marked thyroid hyperplasia. In addition, experimental animals with chronically increased TSH levels caused by antithyroid drugs develop thyroid hyperplasia, adenomas, and carcinomas, and cancer formation is enhanced by additional treatment with a mutagen.23,24 Chronic TSH stimulation apparently provides a “fertile ground” for mutagenesis to occur, but it is likely not a mutagen by itself and relies on other genotoxic events, such as radiation or chemicals, to produce a mutation that would initiate the carcinogenesis.20
The role of hormonal and reproductive factors is not fully understood, despite a significant female predominance among patients with follicular carcinomas. A pooled analysis of 14 case-control studies revealed an increased relative risk of follicular carcinoma in women with artificially induced menopause and in those who miscarried during their first pregnancy, although no specific association with estrogens was found.25 More recently, it has been shown experimentally that circulating estrogens were directly responsible for the increased proliferation rate of thyroid cells in mice that developed thyroid follicular carcinomas as a result of loss of function of the PTEN gene.19 These data provide strong evidence for the role of estrogens in thyroid follicular carcinogenesis in a setting of PTEN loss, although it remains unclear whether this influence remains in human thyroid cells and when PTEN function is preserved.
Familial forms of follicular carcinoma are rare. Some are found in patients with PHTS, which encompasses Cowden syndrome, Bannayan-Riley-Ruvalcaba syndrome, and other rare syndromes caused by a germ line mutation of the PTEN gene located on chromosome 10q23.31. Cowden syndrome is characterized by multiple mucocutaneous hamartomas and a predisposition to malignant tumors, mostly of breast and thyroid. Thyroid lesions are seen in two-thirds of the affected patients, with 10% to 20% developing follicular carcinomas.26,27,28 Thyroid glands from patients affected by this syndrome have characteristic microscopic appearance discussed later in the chapter. Follicular carcinoma may also arise as a manifestation of Werner syndrome. This rare autosomal recessive disease is caused by germ line mutations in the WRN gene on chromosome 8p11-12. It is characterized by premature aging and a higher rate of various malignant tumors. On the basis of a large cohort of Japanese patients affected by this disease, thyroid cancer occurs in approximately 3% of these patients and is typically a follicular carcinoma.29 Thyroid tumors in patients with Werner syndrome manifest at a younger age when compared with the general population and have a lower female to male ratio. Follicular carcinoma may be a rare manifestation of the Carney complex, an autosomal dominant disorder caused by germ line mutations in PRKARIA and other genes and characterized by spotty skin pigmentations, myxomatosis, and other endocrine and nonendocrine tumors.30,31 Isolated familial follicular carcinomas, that is, tumors that are not a part of the known cancer syndromes, are very uncommon. Only few families with isolated conventional or oncocytic follicular carcinoma have been reported.32 In the United States, a family history of thyroid carcinoma (follicular or papillary) is found in 3.7% of patients diagnosed with conventional follicular carcinoma and in 2% of those diagnosed with oncocytic carcinoma.8
PATHOGENESIS AND MOLECULAR GENETICS
Cytogenetic Abnormalities
Clonal chromosomal changes are detected by conventional cytogenetics in approximately 60% of follicular carcinomas.33 They manifest either as chromosome copy number change or as structural chromosomal abnormalities, such as translocations. Numerical chromosomal changes involve loss and/or gain of whole chromosomes or chromosome arms. Most common are gains of chromosome 7 and losses of chromosomes 8, 11, 17, and 18.33,34,35,36,37 Losses of chromosome arms most frequently affect 3p, 11q, and 13q. Recurrent chromosomal translocations are also found, and those tumors rarely show numerical chromosome changes. A t(2;3) (q13;p25) translocation is known to produce the PAX8/PPARγ fusion.38,39 Single cases of tumors with other translocations involving 3p25, such as t(3;7)(p25;q34) and t(1;3)(p13;p25), have been described.38,40 More recently, a tumor with t(3;7)(p25;q34) was found to have another type of PPARγ fusion, CREB3L2/PPARγ.41 Follicular carcinomas with t(7;18)(p15;q24) and t(6;7)(q16;p15) have been reported, suggesting the presence of an important gene on 7p15.33,34
Using comparative genomic hybridization, chromosomal alterations are detected in 60% to 90% of follicular carcinomas.37,42,43 Gains on 1q, 5p, 7p/7q, 12p/12q, and 16p and losses on 3p, 2p, and 8q are most common. Loss of the entire chromosome 22 or 22q appears to be more frequent in widely invasive tumors.42,44 Oncocytic carcinomas more often show losses on 9q.43,45 By fluorescent in situ hybridization (FISH), gains of chromosome 7 are seen in about half of conventional and oncocytic follicular carcinomas.46,47 FISH can also be used for the detection of PAX8/PPARγ rearrangement (Fig. 10.2).39
DNA Ploidy
An aneuploid cell population is found in 50% to 60% of follicular carcinomas and typically correlates with multiple numerical chromosomal changes that are seen cytogenetically.48,49,50 Some, but not all, studies have reported a general tendency of an increased aneuploidy rate from follicular adenomas to follicular carcinomas and from minimally invasive to widely invasive follicular carcinomas.49,50,51
Loss of Heterozygosity
Loss of heterozygosity (LOH) results from a deletion of discrete, small or large chromosomal regions. During tumorigenesis, LOH frequently affects the regions where important tumor suppressor
genes reside. A high rate of LOH is a characteristic feature of follicular carcinomas and of the follicular tumorigenesis in general. A meta-analysis of the data reported in the literature demonstrates a 20% average rate of LOH per chromosome arm in follicular carcinoma, 6% in follicular adenoma, and only 2.5% in papillary carcinoma (Fig. 10.3).52 The different levels of chromosome regions loss signify a fundamental difference in the pathogenesis between follicular and papillary thyroid carcinomas.
genes reside. A high rate of LOH is a characteristic feature of follicular carcinomas and of the follicular tumorigenesis in general. A meta-analysis of the data reported in the literature demonstrates a 20% average rate of LOH per chromosome arm in follicular carcinoma, 6% in follicular adenoma, and only 2.5% in papillary carcinoma (Fig. 10.3).52 The different levels of chromosome regions loss signify a fundamental difference in the pathogenesis between follicular and papillary thyroid carcinomas.
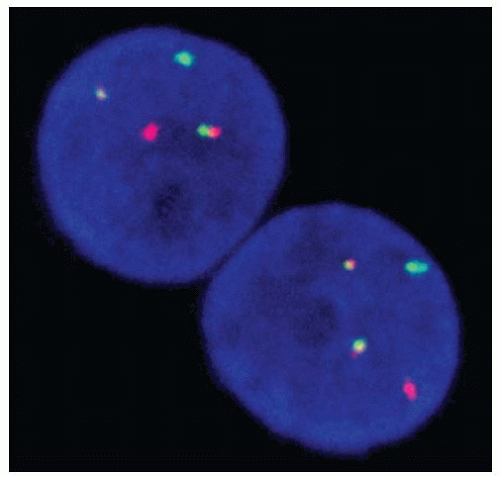 FIGURE 10.2. Detection of PAX8/PPARγ rearrangement in follicular carcinoma cells by FISH. Note that each cell contains one intact PAX8 (red) and one intact PPARγ (green) signals, as well as two fused signal pairs corresponding to the rearrangement between the two gene probes. |
The reported data on LOH in specific chromosomal regions show high variability between individual studies owing to the difference in the locations of microsatellite markers used for each study. Regions on 2p, 3p, 10q, 11p, and 17p were found to be deleted in several studies.36,53,54,55,56 Several of these regions harbor known tumor suppressor genes such as VHL on 3p25-26, TP53 on 17p13, and PTEN on 10q23. Although results vary, investigators have found a higher rate of LOH in oncocytic carcinomas when compared with conventional follicular carcinomas.53,54 LOH may more often affect the regions that are imprinted (i.e., have parent-specific expression of genes) as compared with nonimprinted regions.57
Somatic Mutations
The prevalence of somatic mutations found in follicular carcinoma is summarized in Table 10.2.
RAS Mutations
RAS is the most common mutation known to occur in follicular carcinomas. It is found in almost half of conventional follicular carcinomas and with lower prevalence in oncocytic carcinomas.58,59,60,61 Point mutations involve codons 12, 13, and 61 of the three human RAS genes (HRAS, KRAS, and NRAS). The most frequently affected hot spots are NRAS codon 61 and HRAS codon 61.60,62 These mutations stabilize the protein in its active guanosine triphosphate (GTP)-bound form, leading to constant stimulation of the downstream signaling pathways, particularly the mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3K)/AKT cascades (Fig. 10.4).
RAS mutation is likely to serve as an early and initiating event in thyroid follicular carcinogenesis. However, it may not be sufficient by itself for full malignant transformation. This is supported by occurrence of this mutation not only in carcinomas but also in follicular adenomas. In experiments on cultured cells, the mutant RAS initiates self-limited cell proliferation and promotes chromosome instability, which may predispose the cells for acquiring additional mutations and a more malignant phenotype.63,64 Expression of mutant RAS in transgenic mice leads mostly to the development of hyperplastic nodules and follicular adenomas, whereas additional loss of PTEN results in the rapid development of follicular carcinomas.65,66,67
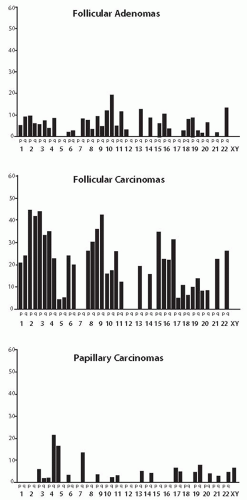 FIGURE 10.3. Rate of LOH per chromosome arm in various types of thyroid tumors. Note a sharp difference in the extent and frequency of LOH between papillary carcinoma and follicular tumors. Reproduced with permission from Ward LS, Brenta G, Medvedovic M, et al. Studies of allelic loss in thyroid tumors reveal major differences in chromosomal instability between papillary and follicular carcinomas. J Clin Endocrinol Metab. 1998;83:525-530. Copyright, The Endocrine Society. |
Table 10.2 Prevalence of Somatic Mutations in Thyroid Follicular Carcinomas | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||
PAX8/PPARγ Rearrangement and Other PPARγFusions
PAX8/PPARγ rearrangement is the second most common genetic alteration in follicular carcinoma. It is found in approximately 35% (range, 0% to 78%) of conventional follicular carcinomas and <5% of oncocytic carcinomas.68 This rearrangement can also be seen in a small proportion (1% to 5%) of the follicular variant papillary carcinomas and in some (˜8%) of follicular adenomas.60,68,69,70,71,72 It is a result of the cytogenetically detectable translocation t(2;3) (q13;p25), which leads to the fusion between the PAX8 gene on 2q13 and the PPARγ gene on 3p25 (Fig. 10.2).39 PAX8/PPARγ-positive tumors tend to present in patients at a younger age and be smaller, and more often show a solid growth pattern and vascular invasion as compared with follicular carcinomas negative for this rearrangement.60,69 Tumors with PAX8/PPARγ almost never show RAS mutations, suggesting that follicular carcinomas develop through two distinct molecular pathways, associated with either PAX8/PPARγ or RAS mutation.60
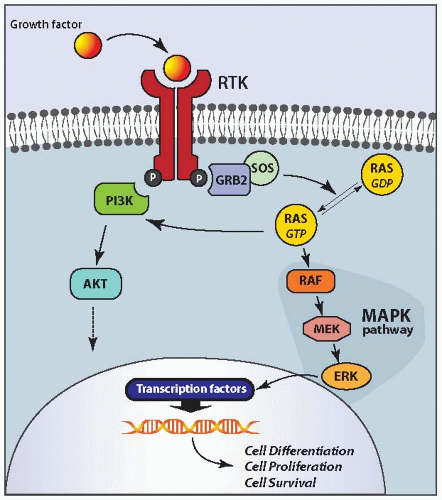 FIGURE 10.4. Molecular pathways activated by mutant RAS in thyroid cells. RAS proteins function as a molecular switch propagating signals from cell membrane receptor tyrosine kinases (RTK) to multiple cytoplasmic targets. On activation, RAS shifts from its inactive, guanosine diphosphate (GDP) bound form to the active, GTP-bound form and phosphorylates downstream cytoplasmic targets before returning to the inactive, RAS-GDP form. Point mutations stabilize the protein in its active form, leading to constitutive stimulation of the RAF-MEK-ERK pathway, also known as MAPK pathway, and PI3K/AKT pathway. |
The PAX8/PPARγ fusion results in strong overexpression of the PPARγ gene. However, the mechanisms of cell transformation induced by this rearrangement are not fully understood. Several proposed possibilities, some of which contradict each other, include inhibition of normal PPARγ function by the chimeric PAX8/PPARγ protein through a dominant negative effect, activation of normal PPARγ targets because of the overexpression of the chimeric protein that contains all functional domains of wild-type PPARγR, deregulation of PAX8 function, and activation of a set of genes unrelated to both wild-type PPARγ and wild-type PAX8 pathways.39,73,74 Some studies that utilized experimental models have observed, in addition to the oncogenic properties, a paradoxical, tumor suppressor effect of this chimeric protein in the form of inhibition of angiogenesis.75,76
In addition to follicular carcinomas, PAX8/PPARγ is found in about 8% of follicular adenomas.68 Although some interpret these data as evidence that the signaling from PAX8/PPARγ is insufficient for the development of malignancy, it is more likely that follicular adenomas positive for this rearrangement are in fact preinvasive or in situ follicular carcinomas, or tumors where invasion was overlooked or not sampled during examination. In support of this possibility, most of PAX8/PPARγ-positive follicular adenomas are highly cellular tumors that show a microfollicular or solid/trabecular growth pattern, have a thick capsule, and express immunohistochemical markers of malignancy such as galectin-3 and/or HBME-1.60,71,77
In addition to PAX8/PPARγ, the PPARγ gene can be fused to other partners. Another fusion partner is CREB3L2; however, this type of PPARγ fusion is rare and has been reported in 1 out of 42 (2.4%) follicular carcinomas in one series.41
PI3K/PTEN/AKT Pathway Mutations
The PI3K/PTEN/AKT signaling pathway plays an important role in the regulation of cell survival, proliferation, and migration.78 In thyroid cancer, the activation of this pathway may be more important for the development and progression of follicular carcinomas than papillary carcinomas.79
In follicular carcinomas, this pathway can be chronically stimulated by activating mutation in the RAS or PIK3CA gene or by inactivating mutation in the PTEN gene (Fig. 10.5). The activation of this pathway induces thyroid cell proliferation and other effects, which particularly rely on the activation of the downstream mammalian target of rapamycin (mTOR) protein.80
The PIK3CA gene, which codes for a catalytic subunit (p110) of PI3K, is mutated in 6% to 13% of follicular carcinomas.81,82,83 The mutations are typically located in exon 20 and exon 9 of the PIK3CA gene that code for the kinase domain and helical domain, respectively. In addition, PIK3CA gene copy number gains are found in up to 25% of follicular carcinomas.84 However, whether the gain of several gene copies is functionally sufficient for the activation of this pathway remains unclear.
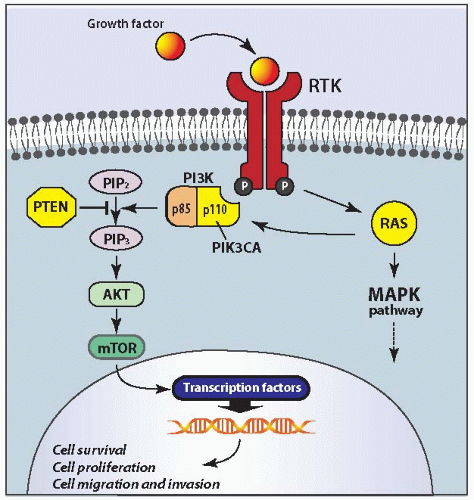 FIGURE 10.5. PI3K/PTEN/AKT signaling pathway and location of mutations in thyroid follicular carcinoma. This pathway transmits signals from cell membrane growth factor receptors (RTK), which activate PI3K directly or through RAS. AKT is activated downstream of PI3K and regulates multiple genes that oppose apoptosis and promote cell survival, proliferation, and migration/invasion. PTEN negatively regulates PI3K signaling and prevents AKT activation. PI3K consists of two subunits, one of which, p110, is coded by the PIK3CA gene. In thyroid follicular carcinomas, this pathway can be activated by gain-offunction mutations of RAS and PIK3CA and loss-of-function mutations of PTEN. PIP2, phosphatidylinositol-3,4-diphosphate; PIP3, phosphatidylinositol-3,4,5-triphosphate. |
Somatic mutations of the PTEN tumor suppressor gene are identified in 6% to 12% of follicular carcinomas.82,83,85 They are point mutations or small frameshift deletions that most frequently affect exons 5 and 7 of the gene. These alterations lead to a loss of function of the PTEN protein, which results in the activation of AKT, mTOR, and other downstream targets. It appears that mutations of RAS, PTEN, and PIK3CA are rarely found in combination in the same tumor.82 This suggests that one of these mutations may be sufficient for the activation of the PI3K/PTEN/AKT pathway. However, the experimental studies have demonstrated that, although mice with thyroidspecific expression of mutant RAS at physiologic levels and mice with loss of PTEN do not develop follicular carcinomas, the double-mutant mice quickly develop invasive and metastatic follicular carcinomas.67 The mice carrying both RAS and PTEN mutations show strong activation of both PI3K and MAPK signaling pathways, which was probably responsible for the accelerated follicular carcinogenesis.
Mitochondria-Related Mutations in Oncocytic Carcinomas
Oncocytic follicular carcinomas are composed of cells with distinct cytoplasmic granularity because of the accumulation of frequently abnormal mitochondria. The cause of the mitochondrial changes and their relationship to the neoplastic process are not fully understood. It is likely that they may represent either a primary change induced by a tumor initiation mutation or a secondary, phenotypic change.2,86,87,88
Mutations of the GRIM-19 gene have been recently identified in oncocytic thyroid tumors.89 This gene encodes a protein that regulates cell death, promotes apoptosis, and affects mitochondrial metabolism, serving as an essential component of complex I of the mitochondrial respiratory chain.90 In one study, somatic missense mutations in GRIM-19 were found in 1 out of 11 of oncocytic follicular carcinomas and in 2 out of 10 of oncocytic variants of
papillary carcinoma.89 These mutations may disrupt the function of this antiapoptotic tumor suppressor gene and promote tumorigenesis. However, the prevalence of GRIM-19 mutations in oncocytic follicular carcinomas and their role in carcinogenesis have yet to be fully established.
papillary carcinoma.89 These mutations may disrupt the function of this antiapoptotic tumor suppressor gene and promote tumorigenesis. However, the prevalence of GRIM-19 mutations in oncocytic follicular carcinomas and their role in carcinogenesis have yet to be fully established.
Mutations in mitochondrial DNA (mtDNA) are found with a high frequency in oncocytic carcinomas.91,92 These mutations, which include deletions and frameshift and missense point mutations, predominantly affect the mitochondrial complex I genes and are predicted to disrupt the gene function. Whether they play the causative role in tumor initiation remains unclear because mtDNA generally has a high mutation rate and these mutations also occur in nonneoplastic oncocytic cells and in nononcocytic tumors.
Alterations in Gene Expression Profiles
High-density cDNA microarrays were used in several studies to identify alterations in gene expression profiles in follicular carcinomas. These studies have identified a large number of genes in which expression was dysregulated in follicular carcinomas as compared with normal thyroid tissue and with follicular adenomas.73,93,94,95,96 Many of these genes are involved in the regulation of cellular growth and proliferation, programmed cell death, thyroid differentiation, development, and cell migration. However, there is little overlap between specific genes found to be differentially upregulated or downregulated in different studies, which makes it difficult to define the role of alteration in the expression of specific genes and follicular carcinogenesis.
PATHOGENETIC RELATIONSHIP BETWEEN FOLLICULAR ADENOMA AND FOLLICULAR CARCINOMA
The relationship between follicular adenoma and follicular carcinoma is not fully understood. Several lines of evidence suggest that these tumors are related biologic entities and point toward the existence of the adenoma to carcinoma progression.
The two tumor types have similar histopathologic appearance in all aspects except for the presence of invasion.
Follicular carcinomas are rarely very small. Only 3% to 8% of follicular carcinomas are ≤ 1 cm as opposed to 27% to 45% of papillary carcinomas.1,4,8 This suggests that many follicular carcinomas may develop from preexisting lesions rather than arise de novo.
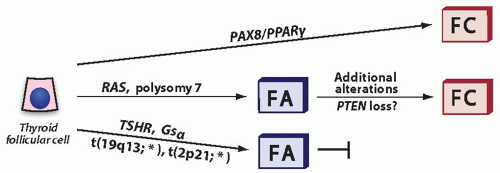
FIGURE 10.6. Putative pathways in the development of follicular tumors and progression from follicular adenoma to follicular carcinoma. Some carcinomas are likely to arise from preexisting adenomas that carry RAS mutation or numerical chromosomal alterations, most frequent of which is chromosome 7 polysomy. Additional alterations are likely required for malignant transformation, possibly loss of function of PTEN. Other carcinomas, such as those harboring PAX8/PPARγ rearrangement, may develop directly, bypassing the adenoma stage. Finally, some adenomas, such as those with translocations involving 19q13 and 2p21 and hyperfunctioning adenomas with TSHR and Gsα mutations, lack the propensity to progress to carcinomas. *These translocations involve fusions to several different partners. FC, follicular carcinoma; FA, follicular adenoma.
Follicular carcinomas typically present at a patient age 8 to 10 years older than adenomas and are larger in size than adenomas, implying a progression over time.26,88,97,98
Experimental mice carrying mutations in the RAS, PTEN, and PRKARIA genes develop first follicular adenomas and then develop follicular carcinomas at an older age or after additional goitrogen stimulation.19,65,66,99,100 Similarly, dietary iodine deficiency in experimental animals leads first to follicular adenoma and after longer intervals to follicular carcinoma.15
Both tumor types share similar molecular and cytogenetic alterations. Mutations of RAS are common, and mutations of PTEN and PIK3CA occur in both tumor types but with higher frequency in carcinomas. Chromosomal gains and losses are frequent and show similar patterns, with polysomy 7 being the most common cytogenetic change in adenomas and carcinomas. Considerable similarity in the comparative genomic hybridization (CGH) profiles also exists.37,43 Both tumor types reveal substantial rates and similar profiles of LOH, but the overall frequency of chromosome arm loss is significantly higher in carcinomas,52 supporting the progression.
Other data suggest that progression from adenoma to carcinoma is not a paradigm of follicular carcinogenesis.
Balanced translocations involving chromosomal loci on 19q13 or 2p21 are common in adenomas but are exceptionally rare in carcinomas, suggesting that adenomas harboring these translocations have low potential for progression to carcinomas. Similarly, hyperfunctioning (“toxic”) thyroid adenomas caused by mutations in the TSHR and Gsα genes (see Chapter 8) exceedingly rarely progress to follicular carcinomas.
PAX8/PPARγ rearrangement occurs in a significant portion of follicular carcinomas but is rarely found in adenomas, suggesting that tumors with this genetic alteration develop without a preexisting stage of adenoma.
These findings provide evidence, although indirect, for the existence of three different pathways in thyroid follicular tumorigenesis (Fig. 10.6).
CLINICAL PRESENTATION AND IMAGING
Follicular carcinomas typically present as a slowly enlarging, painless, solitary thyroid nodule. Patients are usually asymptomatic, although dysphagia, hoarseness, and/or stridor may
sometimes accompany the mass. In rare cases, the presenting symptoms are caused by distant metastases, such as bone pain or pathologic bone fracture. On physical examination, the nodule typically moves with swallowing. Nodules that are fixed to surrounding tissues raise strong suspicion for invasive malignancy. Thyroid function tests are normal. Radionuclide scans typically reveal a “cold” nodule because follicular carcinomas concentrate radioiodine or other tracers less avidly than adjacent thyroid parenchyma. Although single cases of “hot,” hyperfunctioning follicular carcinomas of both conventional and oncocytic type have been described,101,102,103 they are exceedingly rare. On ultrasound examination, tumors usually appear as a solid hypoechoic nodule with a peripheral halo, the latter representing the fibrous capsule (Fig. 10.7). The outlines of the nodule are well defined and smooth, unless the tumor is widely invasive. Ultrasound features do not reliably differentiate follicular carcinomas from adenomas. However, the presence of irregular, poorly defined margins, a thick irregular capsule, and chaotic intranodular blood flow on color Doppler imaging is more suggestive of carcinoma. A routine chest x-ray that includes the neck may reveal tracheal deviation and pulmonary metastases. CT and MRI can be used when invasion is suspected to better evaluate the extension of tumor into the adjacent neck structures. Fine needle aspiration (FNA) is routinely performed on solitary thyroid nodules. Because FNA cytology typically reveals a cellular aspirate suggestive of a follicular tumor, the patients are referred for surgery.
sometimes accompany the mass. In rare cases, the presenting symptoms are caused by distant metastases, such as bone pain or pathologic bone fracture. On physical examination, the nodule typically moves with swallowing. Nodules that are fixed to surrounding tissues raise strong suspicion for invasive malignancy. Thyroid function tests are normal. Radionuclide scans typically reveal a “cold” nodule because follicular carcinomas concentrate radioiodine or other tracers less avidly than adjacent thyroid parenchyma. Although single cases of “hot,” hyperfunctioning follicular carcinomas of both conventional and oncocytic type have been described,101,102,103 they are exceedingly rare. On ultrasound examination, tumors usually appear as a solid hypoechoic nodule with a peripheral halo, the latter representing the fibrous capsule (Fig. 10.7). The outlines of the nodule are well defined and smooth, unless the tumor is widely invasive. Ultrasound features do not reliably differentiate follicular carcinomas from adenomas. However, the presence of irregular, poorly defined margins, a thick irregular capsule, and chaotic intranodular blood flow on color Doppler imaging is more suggestive of carcinoma. A routine chest x-ray that includes the neck may reveal tracheal deviation and pulmonary metastases. CT and MRI can be used when invasion is suspected to better evaluate the extension of tumor into the adjacent neck structures. Fine needle aspiration (FNA) is routinely performed on solitary thyroid nodules. Because FNA cytology typically reveals a cellular aspirate suggestive of a follicular tumor, the patients are referred for surgery.
GROSS FEATURES
On gross examination, follicular carcinomas typically appear as an encapsulated nodule of oval or round shape (Fig. 10.8A). Most are 2 to 4 cm in size, although they can be larger. The cut surface is solid and fleshy, and in fresh, nonfixed samples, the tumor is frequently seen bulging from the confines of the capsule. The color of the nodule is gray-white in conventional type carcinomas and brown-tan or mahogany-brown in oncocytic carcinomas. A thick fibrotic capsule frequently surrounds the tumor. A very thick capsule is more suggestive of malignancy. Invasion of the capsule is rarely found on gross examination, although it can be identified in some cases (Fig. 10.8A). Widely invasive tumors have widespread invasion through the capsule, and in some cases, no residual capsule is recognized (Fig. 10.9). Foci of hemorrhage, infarction, and other secondary changes, either spontaneous or FNA induced, may be seen.
Macroscopic evaluation of an encapsulated solitary nodule requires careful inspection of the capsule to identify areas of invasion. Unless the nodule is very large, the entire pericapsular zone should to be submitted for microscopic examination. If this is not possible, at least 10 sections from the capsule should be processed because the chance of detecting invasion progressively increases when 1 to 10 blocks are examined.104 The details of gross examination are described in Chapter 19.
Most follicular carcinomas present as a solitary nodule in the background of normal appearing thyroid parenchyma, although macroscopic changes suggestive of lymphocytic thyroiditis or nodular hyperplasia may also be seen in the adjacent thyroid tissue. The presence of multiple well-defined solid thyroid nodules, particularly in a young patient, should raise the possibility of an inherited disease, such as PHTS, as discussed later in the chapter.
MICROSCOPIC FEATURES
Follicular carcinomas typically have a well-formed and complete capsule, which is thick (0.1 to 0.3 cm) or at least moderately thick (0.1 cm) (Fig. 10.10).105,106 The capsule consists of parallel layers of collagen fibers and frequently contains medium caliber blood vessels; the latter may show an edematous and irregularly thick smooth muscle wall. In rare cases, carcinomas have a thin or incomplete capsule.
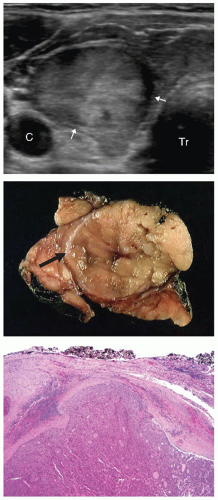 FIGURE 10.7. Follicular carcinoma. Top: An ultrasound image shows a large ovoid nodule with variable echogenicity, slightly irregular outlines, and hypoechoic, focally irregular halo (arrows). Tr, trachea; C, carotid artery. Middle: Gross photograph showing an encapsulated nodule (arrow) with no obvious invasion. Whitish appearance of the adjacent thyroid tissue is because of marked chronic lymphocytic thyroiditis. Bottom: Microscopic image demonstrating capsular invasion, which is diagnostic for follicular carcinoma. |
Architectural patterns of follicular carcinomas are similar to those seen in follicular adenomas, although the proportion of more
cellular growth patterns is higher in carcinomas. A microfollicular or solid/trabecular growth pattern is found in approximately 80% of cases and normofollicular or macrofollicular pattern with high colloid content in approximately 20%.104 A nested or insular architecture may also be seen. Although the growth pattern has no diagnostic value per se, it should alert a pathologist to search more extensively for invasion, especially if the lesion is very cellular. Likewise, the architectural patterns do not correlate with the frequency of metastases or cancer-related death.107,108 Tumors with solid, trabecular, and insular growth patterns should be distinguished from poorly differentiated thyroid carcinoma, which has a significantly more aggressive behavior.
cellular growth patterns is higher in carcinomas. A microfollicular or solid/trabecular growth pattern is found in approximately 80% of cases and normofollicular or macrofollicular pattern with high colloid content in approximately 20%.104 A nested or insular architecture may also be seen. Although the growth pattern has no diagnostic value per se, it should alert a pathologist to search more extensively for invasion, especially if the lesion is very cellular. Likewise, the architectural patterns do not correlate with the frequency of metastases or cancer-related death.107,108 Tumors with solid, trabecular, and insular growth patterns should be distinguished from poorly differentiated thyroid carcinoma, which has a significantly more aggressive behavior.
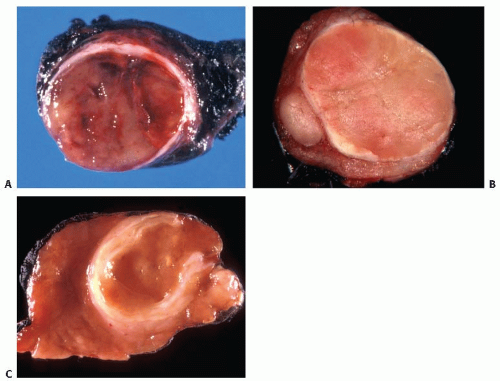 FIGURE 10.8. Gross appearance of follicular carcinoma. A: This follicular carcinoma of conventional type shows a moderately thick capsule and solid cut surface with foci of hemorrhage. The gross appearance is indistinguishable from that of follicular adenoma. B: This follicular carcinoma of conventional type shows a well-defined moderately thick capsule with a focus of tumor located outside of the capsule. C: This follicular carcinoma of oncocytic type shows characteristic mahogany-brown cut surface and a very thick capsule with a clearly identifiable focus of tumor invasion through the capsule. |
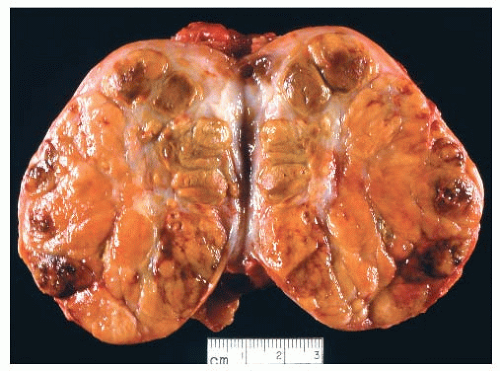 FIGURE 10.9. Widely invasive follicular carcinoma of oncocytic type. The tumor replaces the entire lobe of the thyroid and shows multifocal hemorrhage and necrosis. |
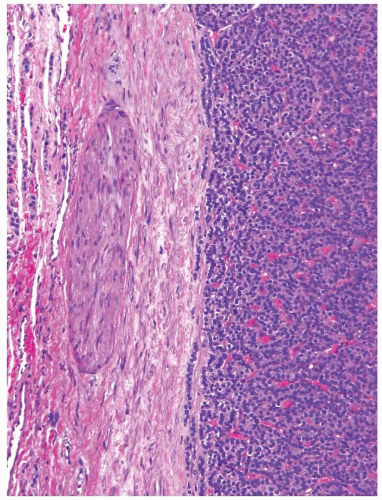 FIGURE 10.10. Low-power view of follicular adenoma. Note a thick fibrous capsule containing a medium-sized artery. |
The cells are typically cuboidal (Fig. 10.11). The cytoplasm is moderately abundant, light eosinophilic to amphophilic. The nuclei are usually small to medium and round, with smooth contours and dark or more vesicular chromatin. Small nucleoli are frequently seen in conventional type carcinomas but are much more prominent in oncocytic follicular carcinoma. Some tumors have more irregular nuclei and coarse chromatin. The irregular nuclear contours raise suspicion for papillary carcinoma, but other nuclear features of papillary carcinoma are lacking (Fig. 10.12A). Rarely, random cells with very large, hyperchromatic, and highly irregular nuclei may be found (Fig. 10.12B). This exuberant nuclear atypia of single cells does not increase the likelihood of malignancy in thyroid tumors, as it is also seen in follicular adenomas and hyperplastic nodules.
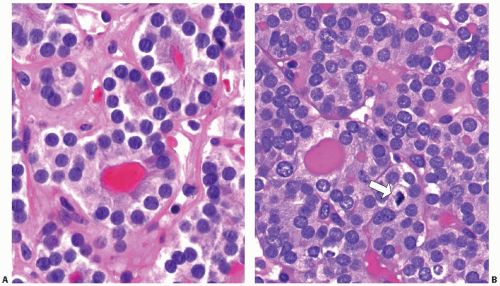 FIGURE 10.11. High-power views of two follicular carcinomas. A: This tumor shows cells with uniform, dark nuclei with smooth contours. B: Cells of this tumor have more atypical, enlarged nuclei with coarse chromatin and significant variability in nuclear size and shape. A mitotic figure is seen (arrow). |
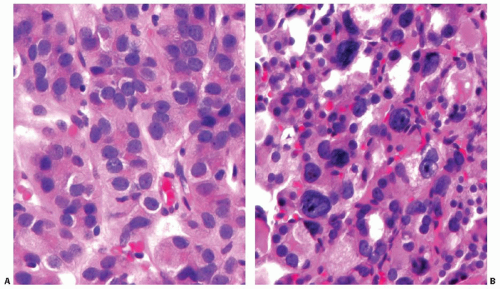 FIGURE 10.12. Appearance of nuclei in follicular carcinoma cells. A: Nuclei of this follicular carcinoma have substantial irregularity of contours, although overall the nuclear features are insufficient to classify this tumor as papillary carcinoma. B: Cells with highly atypical hyperchromatic nuclei are seen in this follicular carcinoma. The presence of scattered bizarre cells does not constitute evidence for malignancy. |
Mitotic figures may be found, typically 1 to 2 per 10 high-power fields (Fig. 10.11B). Higher mitotic activity may mark the
areas of recent aspiration of other secondary changes. Atypical mitoses are rare. Well-differentiated follicular carcinomas do not show tumor necrosis unless seen in association with spontaneous or FNA-induced secondary changes. When present, it suggests the emergence of poorly differentiated thyroid carcinoma.109
areas of recent aspiration of other secondary changes. Atypical mitoses are rare. Well-differentiated follicular carcinomas do not show tumor necrosis unless seen in association with spontaneous or FNA-induced secondary changes. When present, it suggests the emergence of poorly differentiated thyroid carcinoma.109
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


