Focal Segmental Glomerulosclerosis Classification
A. Brad Farris, III, MD
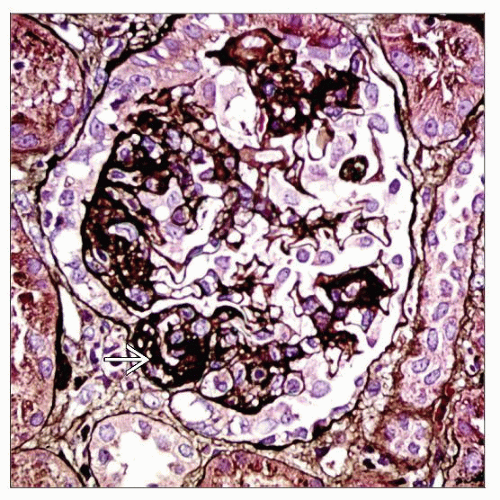 Light microscopy of a case of collapsing FSGS shows a glomerulus with areas of sclerosis  (silver stain). (silver stain). |
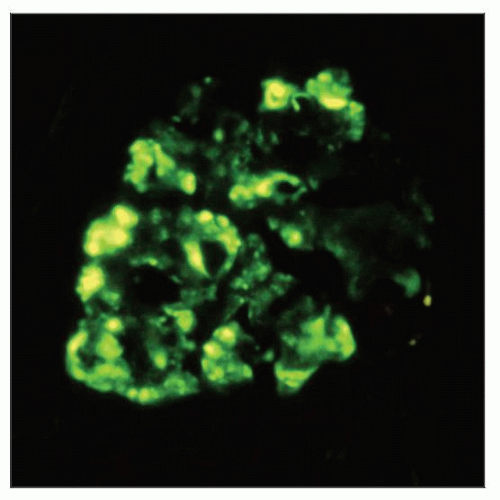 Immunofluorescence for C3 shows a predominantly segmental pattern of staining in the mesangium and in areas of glomerular capillary loop collapse in a case of FSGS. |
TERMINOLOGY
Abbreviations
Focal segmental glomerulosclerosis (FSGS)
Definitions
Group of podocytopathies (of varied etiology) that share the feature of focal segmental glomerulosclerosis, typically with moderate to heavy proteinuria
CLASSIFICATION SYSTEMS
Morphologic Classification
Based on glomerular morphology categories defined by the Columbia classification
Collapsing variant
Glomerular tip lesion
Cellular variant
Perihilar variant
FSGS not otherwise specified (NOS)
Etiologic Classification
Based on identified causes of FSGS: Some correspondence with morphologic variants
Familial/genetic
Increasing number of genes identified
β-integrin mutations
α-actinin-4 mutations
WT-1 mutations
Podocin mutations
Drug-induced
Pamidronate
Heroin (heroin nephropathy)
Lithium
Interferon-α and -β
Virus
HIV (HIV-associated nephropathy [HIVAN])
Parvovirus B19
Adaptive, structural-functional responses
Secondary to congenital or acquired reduction in nephrons relative to body mass
Oligomeganephronia, unilateral renal agenesis, renal dysplasia, reflux nephropathy, hypertension, anabolic steroids
Vascular
Hypertension
Thrombotic microangiopathy
Atheromatous emboli
Calcineurin inhibitor toxicity
Idiopathic
Usual primary form, either NOS or collapsing variant
Plasma factor responsible, identity sought
EPIDEMIOLOGY
Incidence
Most common cause of nephrotic syndrome in adults
Apparent increased incidence over past 2 decades
˜ 25% of adult nephropathies, compared with < 10% 20 years ago
History
Focal and segmental glomerular hyalinization and capillary loop degradation described by Fahr in 1925
Arnold Rich described juxtamedullary glomerulosclerosis in children dying with nephrotic syndrome in 1957
FSGS was recognized as a distinct entity by International Study of Kidney Diseases in Children in the 1970s
Collapsing variant was recognized by Mark Weiss in the 1980s and later as usual pattern of HIVAN
ETIOLOGY/PATHOGENESIS
Pathogenesis
Now classified as podocytopathies
Familial podocyte protein defects
Mutations in TRPC6, a calcium-permeable cation channel, lead to abnormal podocyte function and hereditary FSGS
FSGS with defects in α-actinin-4 (ACTN4), podocin (NPHS2, defective in corticosteroid-resistant nephrotic syndrome), and nephrin (NPHS1, defective in congenital nephrotic syndrome of the Finnish type) are relatively rare familial forms of FSGS
Nephrin interacts with CD2-associated protein (CD2AP), and podocin interacts with the nephrin-CD2AP complex
Mutations in WT1 transcription factor, which regulates several podocyte genes, lead to FSGS syndromes (Denys-Drash and Frasier syndromes)
Abnormal cytokines are now thought to play major role in idiopathic FSGS development
Circulating factor identified in patients who have recurrent FSGS after transplant, typically occurring within months after transplantation
Podocyte dysregulation/dysfunction
Differentiation markers of podocytes (e.g., Wilms tumor WT-1 protein, podocalyxin, and synaptopodin) disappear in collapsing FSGS and HIVAN
Loss of podocytes leads to adhesions
Risk factor in African descent is APOL1 gene
Relationship with minimal change disease (MCD) unclear
Dystroglycan, an integral component of the GBM, is decreased in MCD but maintained in nonsclerotic segments in FSGS
CLINICAL IMPLICATIONS
Prognosis
Generally poor with substantial fraction progressing to end-stage renal disease
Clinical Presentation
Proteinuria
Nephrotic syndrome
Azotemia
Treatment
Plasmapheresis has been used to induce remission in some patients with recurrent FSGS
MACROSCOPIC FINDINGS
General Features
Pale yellow kidneys due to lipid in tubules
MICROSCOPIC FINDINGS
General Features
Glomeruli
Sclerosis involves some glomeruli (focal) and only a portion of glomerular tuft (segmental)
Diagnosed even when only 1 glomerulus involved
Global sclerosis may be an incidental finding and is not particularly useful in making diagnosis of FSGS
Adhesions (synechiae) of glomerular tuft to Bowman space often accompany segmental sclerosis and are often seen early in sclerosis process
Hyalinosis
Portion of glomerular involvement has a smooth, glassy (hyaline) appearance
Typically thought to occur from insudation of plasma proteins
Increased matrix with obliteration of glomerular capillary lumen
FSGS has zonal distribution, beginning in corticomedullary (juxtamedullary) junction (CMJ)
Important to note whether sampling of CMJ is included
Glomerular hypertrophy often accompanies FSGS
Potential surrogate marker in cases without sampled segmental sclerosis
Tubules
Tubular epithelial cells contain PAS(+) reabsorption droplets due to glomerular proteinuria
Tubular atrophy is typically only focal early in course of FSGS
TBM thickened in areas of atrophy
Tubular atrophy may be more prominent late in course of the disease
Tubulointerstitial changes pronounced in collapsing variants of FSGS and in HIVAN
Cystic dilatation and more prominent lymphoid infiltrate
Interstitium
Interstitial fibrosis (IF) may be present and is typically focal
Some cases have extensive IF ± tubular atrophy (TA), and IF/TA in young patients may indicate unsampled FSGS in patients without identified glomerulosclerosis
Interstitial inflammation is absent or minimal
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


