Fig. 1
Signals detected by ratio imaging and FilterFRET. (a–c) Spectra of a perfect FRET pair, i.e., there is no leak-through and cross-excitation between the fluorophores. (d–f) Spectra of real-world fluorophores—the CFP and YFP pair. Note that there is considerable leak-through of the emission of each fluorophore into the detection range of its partner. Acceptor molecules can be cross-excited at the donor excitation wavelength, but the excitation of donor molecules with an acceptor excitation wavelength is always negligible. (a, d) Excitation and emission spectra of a FRET pair with optimized excitation wavelengths (λ d exc and λ a exc) and detection ranges (λ d em and λ a em) indicated (a, perfect FRET pair; b, real-world fluorophores). (b, e) Signal detected in d and s channels with FRET (dotted line) or without (continuous line). Note that signals in d and s channels are detected after exciting with donor excitation wavelength (λ d exc), (b, perfect FRET pair; e, real-world fluorophores). (c, f) Signal detected in channel a (with an excitation optimized for acceptor molecules—λ a exc) is not affected by FRET (c, perfect FRETpair; f, real-world fluorophores). (g) Table summarizing signals present in the channels of a FRET experiment. Note that in case of a perfect FRET pair only diagonal elements are nonzero. Non-diagonal signals are present only in case of leak-through (signals marked with *), cross-excitation (signals marked with **), or both (signals marked with * **). Donor molecules are essentially not cross-excited at acceptor excitation wavelength (I a D − S and I a S signals are grayed out). Equations to the right of the table introduce parameters linking the signals, originating from the same population of molecules but detected in different channels
d—donor excitation (λ d exc) and donor emission (λ d em).
s—donor excitation (λ d exc) and acceptor emission (λ a em).
a—acceptor excitation (λ a exc) and acceptor emission (λ a em).
The fluorophores in this thought experiment are designed to be perfect for FRET experiments (Fig. 1a). A huge overlap between the emission spectrum of the donor and the absorption spectrum of the acceptor maximizes the possible efficiency of FRET. Yet there is neither cross-excitation between the fluorophores nor leak-through of fluorescence emission of one of them into the channel optimum for its partner.
The signal of the non-interacting molecules in this experiment can be detected directly in channel d (Fig. 1b continuous line) and channel a (Fig. 1c). If molecules of interest can interact with each other, we expect FRET between their fluorophores. The signal in channel d is diminished (I D − S ) as now the donors that are in a complex transfer part of their absorbed energy to the acceptor molecule, rather than emitting it directly as photons. Signal of acceptors excited by FRET (sensitized emission, I S ) appears in channel s. The signal detected in channel a does not change as the presence of FRET does not interfere with the signal of (directly) excited acceptor molecules—I A (Fig. 1c). Thus, we have introduced three detection channels as well as three populations of molecules. This setup is summarized in the table (Fig. 1g). In case of a perfect FRET pair, only diagonal elements of the table are nonzero—i.e., the signal from different populations of molecules can be detected only in the channels optimized for them. It is the signal of sensitized emission detected in channel s that provides the information on the presence and magnitude of sensitized emission and as a consequence about the presence of interaction between the studied molecules.
Unfortunately, in the experiments performed with real-world fluorophores, cross-excitation and leak-through usually complicate matters (Fig. 1d). The signal in channel s is no longer a measurement of pure sensitized emission. With (Fig. 1e dotted line) or without FRET (Fig. 1e continuous line), it always contains a leak-through signal from donor molecules and a signal from cross-excited acceptor molecules. In this situation, some signal from any of the three populations of molecules can be detected in all three detection channels (Fig. 1g). For example, Hence the signal in channel d (first column in the table Fig. 1g) is the sum of the signal from excited non-fretting donor molecules (I D − S d ), signal of sensitized emission leaking back into the donor channel (I S d ), and signal of cross-excited acceptor molecules also leaking back into the donor channel (I A d ).
In most experiments, cross-excitation of donor molecules is very small and can be neglected. Hence the signal in channel a originates only from the population of acceptor molecules—I A a (I D − S a and I S a appear gray in the table in Fig. 1g). Nevertheless, the measurements obtained from the channels d, s, and a do not suffice to calculate the value of the sensitized emission signal (I S s ) because we have to determine seven different intensities (non-gray in the table) from just three measurements. Note however that in any specific row in the table (Fig. 1g), all the intensities originate from a single population of molecules. Therefore, a set of parameters (α, β, γ, and δ) that relate them to each other can be introduced (see equations to the right of the table in Fig. 1g). These parameters depend solely on the specific settings of the microscope (filters, spectral sensitivity, etc.) and on the spectral characteristics of donor (β) or acceptor (α, γ, and δ) molecules. Therefore, they can be calculated based on measurements of pure populations of donors or acceptors. For example, β just represents the fraction of leak-through of donors in the s channel.
On wide-field fluorescence microscopes, these factors are fixed for a given fluorophore. However, the parameters of the excitation and detection pathways in a confocal microscope can be tuned independently. Moreover, the intrinsic instability of laser intensities may easily amount to 5 %, even in modern instruments, making it often impossible to recreate exactly identical settings between the experiments. As a consequence, it is best to collect the data on the magnitude of the correction factors at the same time as performing the FRET experiment.
Based on the measurement of three channels together with four introduced parameters, it is finally possible to solve the system of seven equations to calculate the value of sensitized emission, I S s :
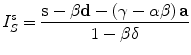 It is crucial to understand what kind of information is provided by the sensitized emission signal. By definition, this signal is a measurement of the amount of energy being transferred. It follows the spatial distribution of interactions between proteins or their activity and depends on the number of fluorophores available to interact with each other. Note that because sensitized emission is sensitive to the parameters of both excitation and emission, it is not possible to quantitatively compare two experiments as long as they are not acquired in exactly the same conditions.
It is crucial to understand what kind of information is provided by the sensitized emission signal. By definition, this signal is a measurement of the amount of energy being transferred. It follows the spatial distribution of interactions between proteins or their activity and depends on the number of fluorophores available to interact with each other. Note that because sensitized emission is sensitive to the parameters of both excitation and emission, it is not possible to quantitatively compare two experiments as long as they are not acquired in exactly the same conditions.
FRET efficiency, on the other hand, is fully quantitative and can thus be compared between different experiments. Whether it is necessary to calculate FRET efficiency depends on the nature of the biological question that underlies the experiments. To calculate FRET efficiency, the measured sensitized emission has to be normalized, e.g., by dividing it by the intensity of the (unquenched) donor signal (E d), signal of acceptors (E a), or a combination of the two of them (see Note 1 ). Such normalizations are never straightforward. For example, to determine E d, channels d and s are used that share the same excitation parameters but differ in sensitivity to donors and acceptors, respectively. On the other hand, the channels a and s that are needed to calculate E a may have the same detections settings but do not share the same excitation intensities. Thus, quantitative determination of FRET efficiency requires an additional calibration, the details of which are beyond the scope of this chapter but may be found in literature [11].
The first protocol in the method section of this chapter describes a confocal experiment designed to study the interactions between nexin proteins (SNX1) by measuring FRET. Overexpression of a fusion construct (SNX with fluorescent proteins) enables the detection of signal mainly from tubulovesicular endosomal membranes and from the cytoplasm [12]. Therefore, confocal microscopy was chosen to visualize the fine tubular structures by optical sectioning. In this experiment, we make use of reference cells, expressing only one fluorophore, to calculate the values of the correction parameters.
The second protocol in the method section shows how to measure the intracellular concentration of cAMP with an EPAC-based sensor. For this single-polypeptide FRET sensor, ratio imaging is the method of choice. Ratio imaging, i.e., simply recording the ratio of the d and s images, is the easiest and most popular technique of FRET microscopy. The ratio image is insensitive to fluctuations in excitation light intensity and it normalizes signal differences caused by cell morphology (e.g., differences in cell height) and in expression level. If the stoichiometry of donor and acceptor fluorophores is fixed, quantitative data can be obtained simply by performing an end-point calibration. This can, for example, be the addition of forskolin together with IBMX which drives cAMP levels so high that they saturate the response of the FRET sensor.
2 Materials
2.1 Cell Culture
1.
Cell culture media.
(a)
HEK293 (Embryonal Kidney Cells, American Type Culture Collection), N1E-115 (mouse neuroblastoma), and HeLa (cervical cancer) were cultured in DMEM (Gibco) supplemented with 8 % FCS (Sigma) and 1 % antibiotics (penicillin and streptomycin), in a 5 % CO2, 37 °C, humidified incubator.
(b)
Cells were imaged in DMEM-F12 medium (Gibco).
(c)
Transfection reagent—PEI (Polyethylenimine, Polysciences) was dissolved in ethanol to the concentration of 1.5 mg/ml and stored in a glass container in −20 °C.
2.
Agonists were added from 1,000-fold concentrated stocks:
(a)
Adrenalin (Sigma).
(b)
Forskolin (Santa Cruz Biotechnology).
(c)
IBMX (Sigma).
3.
Coverslips (Thermo Scientific, 24 mm, #1) were sterilized in 70 % ethanol. Each coverslip was put in a separate well of a 6-well plate and left in a laminar flow to dry.
4.
Plasmids.
(a)
SNX1-CFP and SNX1-YFP constructs were a kind gift of Professor Peter Cullen, University of Bristol, England.
2.2 Microscopy and Image Analysis
2.2.1 Sensitized Emission
1.
A confocal microscope capable of exciting CFP (e.g., with a 442 nm HeCd laser line or a 458 nm Argon laser line) and YFP (e.g., a 514 nm Argon laser line). Readout of the signal should be simultaneous with two PMT detectors in the ranges 470–510 and 520–600 nm (slight deviations from these ranges work too). A high-NA immersion objective chromatically corrected for CFP/YFP detection should be used.
2.2.2 Ratiometric FRET
1.
Any inverted fluorescent microscope that can excite CFP (in a range 430–470 nm) and collect donor (470–510 nm) and acceptor (520–600 nm) emission. If a wide-field system is used, it would be beneficial to use an image-splitting device to acquire the images of donor and acceptor simultaneously.
2.
Image acquisition software capable of calculating and plotting the ratio between CFP and YFP emission in a selected region of interest (ROI) during an experiment.
2.2.3 Temperature Control
The imaging systems were equipped with a thermostated chamber for live cell imaging (37 °C) with controlled atmosphere of 5 % CO2 and elevated humidity. If not available, a HEPES-buffered system should be used.
3 Methods
Below we provide two protocols for preparing and performing intensity-based FRET experiments. The first protocol gives detailed information about the use of sensitized emission (FilterFRET) to study interactions of proteins. The second one deals with ratio FRET imaging of dynamic changes of small molecules inside the cells.
3.1 Sensitized Emission/FilterFRET
3.1.1 Planning of the Experiment
1.
Select a pair of fluorescent proteins that constitute a good FRET pair. Their spectral properties have to be compatible with the sample to be studied and the image acquisition system (see Notes 2 and 3 ).
To perform the example experiment, the classical FRET pair CFP (Cyan Fluorescent Protein) and YFP (Yellow Fluorescent Protein) has been chosen.
2.
Prepare fusion constructs of the proteins of interest with fluorescent proteins. Test extensively whether tagging with fluorescent protein may affect the localization or function of the proteins (see Note 4 ).
For this example two constructs SNX1-CFP and SNX1-YFP have been prepared.
3.




Prepare reference cells, i.e., cells that express either donor or acceptor fluorophores alone. Make sure that their signal matches the signal from the cells of interest (see Note 5 ). It is convenient to choose the reference constructs so that the reference cells can be easily discriminated from the FRET cells (see Note 6 ).
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
