(1)
Faculty of Science, Department of Chemistry, National University of Singapore, Singapore, Singapore
(2)
Department of Chemistry, Faculty of Science, National University of Singapore, Singapore, Singapore
(3)
Mechanobiology Institute, Singapore, Singapore
(4)
Faculty of Science, Department of Chemistry and Department of Biological Sciences, National University of Singapore, Singapore, Singapore
Abstract
Fluorescence cross-correlation spectroscopy (FCCS) is a single-molecule sensitive technique to quantitatively study interactions among fluorescently tagged biomolecules. Besides the initial implementation as dual-color FCCS (DC-FCCS), FCCS has several powerful derivatives, including single-wavelength FCCS (SW-FCCS), two-photon FCCS (TP-FCCS), and pulsed interleaved excitation FCCS (PIE-FCCS). However, to apply FCCS successfully, one needs to be familiar with procedures ranging from fluorescent labeling, instrumentation setup and alignment, sample preparation, and data analysis. Here, we describe the procedures to apply FCCS in various biological samples ranging from live cells to in vivo measurements, with the focus on DC-FCCS and SW-FCCS.
Key words
Fluorescence cross-correlation spectroscopy (FCCS)DC-FCCSSW-FCCSTP-FCCSMolecular interactionsSingle molecule spectroscopyQuantitative biologyZebrafish imaging1 Introduction
Fluorescence cross-correlation spectroscopy (FCCS) is an extension of fluorescence correlation spectroscopy (FCS). FCS analyzes the signals from particles labeled with a single fluorescent species and extracts information about concentrations and molecular dynamics. On the other hand, FCCS analyzes the fluorescence signal from two or more particle species that are each tagged with a fluorophore of a distinct emission spectrum. The signals from the distinct fluorophores are recorded in separate wavelength-dependent detection channels, and the signals are cross-correlated to retrieve information about the interaction between the different fluorophore species. Because FCCS relies on the cross-correlation of different signals, it reports directly on molecular interactions and is not dependent on any changes in diffusion coefficients or mass as is FCS [1] or on the interactions between the labels as is the case in Förster resonance energy transfer (FRET) [2]. Therefore, FCCS is particularly useful in quantitating molecular interactions that happen between molecules with similar molecular weight [3] and is complementary to imaging techniques, providing essentially the same spatial resolution but adding information on molecular interactions [4].
FCCS was first demonstrated by J. Rička and Th. Binkert in 1989 using scattered light from nonfluorescent latex particles in the blue channel and fluorescence from labeled latex particles in the green channel [5]. This technique found widespread application when Schwille and colleagues successfully measured DNA-DNA renaturation through detecting and cross-correlating the fluorescently tagged DNA in 1997 using a DC-FCCS setup [6], in which the different dyes are excited with separate lasers. Since then the application of FCCS ranges from the quantitation of biomolecular interactions, protein oligomerizations, to enzyme activity [7–11].
DC-FCCS, while a very powerful technique, suffers also from problems. Initially, the alignment of two lasers to precisely the same spot was difficult; cross talk between the two detection channels and moving structures, e.g., the plasma membrane of living cells, gave rise to a false-positive cross-correlation background. Therefore new FCCS modalities were suggested. Single-wavelength FCCS (SW-FCCS) has the advantage of easier calibration, simpler setup, and elimination of nonideal overlap of excitation volumes by exciting several fluorophores simultaneously using one single laser line [3, 10–12]. Similarly, two-photon excitation FCCS (TP-FCCS) uses a single laser line for the two-photon excitation of two separate fluorophores [13]. Although the counts per particle per second (cps) achieved by TP-FCCS is generally lower than that of DC-FCCS due to photobleaching [14] and saturation [15], TP-FCCS provides potentially a wider selection of fluorophore pairs compared to SW-FCCS [16]. Another derivative, pulsed interleaved excitation FCCS (PIE-FCCS) uses two lasers that are pulsed alternatively to overcome the spectral cross talk among fluorophores. The laser pulses are alternated in the range of nanoseconds such that the emission and excitation cycles of one type of fluorophore are completed before the next fluorophore is excited [17]. PIE-FCCS thus generates crosstalk-free cross-correlations. In addition, it can determine contributions from FRET, which can be used to correct FCCS data [18, 19]. Cell and membrane movement can be corrected by using scanning FCCS [20] (see Note 3).
Here, we describe a DC-FCCS/SW-FCCS procedure starting from sample preparation and emphasizing calibration and measurements which can be used for the study of biomolecular interactions in cells and zebrafish embryos [3, 10, 11, 21, 22]. PIE-FCCS is discussed in another protocol in the same publication.
2 Materials
2.1 Fluorophores for FCCS Experiments
The most important characteristic of fluorophore pairs required for FCCS measurements is the separation of spectra. For DC-FCCS, separated excitation and emission spectra are used for selective excitation and the reduction of cross talk, respectively. For SW-FCCS and TP-FCCS, the fluorophores need to have similar excitation spectra for one- or two-photon excitation, respectively. Besides the excitation and emission spectra, the aspects to be considered include molecular brightness (dependent on extinction coefficient and quantum yield), photostability, and available laser lines [4]. The fluorophores used include organic dyes, quantum dots and nanoparticles, and fluorescent proteins [23] (see Note 4).
2.1.1 Organic Dyes
There is a wide range of commercially available organic dyes with different excitation and emission spectra serving all possible laser lines, labeling a wide range of biological structures, and with very good photophysical properties. The obstacle for the use in live cell and in vivo measurements is the problem of introducing these extrinsic labels specifically and with defined labeling ratios into the sample. Several approaches have been introduced and allow linking the dye to molecules of interests, including synthesized DNA [6], genetically labeled recombinant proteins [24, 25], labeled antibodies [25–28], and extrinsic labeling of proteins through specific reactions with synthetic probes [29, 30]. Besides, pure dye solutions are widely used for system calibration and provide the basis for comparing the performance and measurements of different systems.
2.1.2 Quantum Dots (QDs) and Nanoparticles
QDs are high-quantum-yield fluorescent semiconductor nanoparticles, which can be excited with a single wavelength in the blue region and have size- or composition-dependent emission spectra [31–33]. They are therefore potential probes for SW-FCCS [12], but they can suffer from aggregation making cross-correlation measurements difficult. For FCCS measurements, commercial QD streptavidin conjugates are reported for studying binding between G-protein-coupled receptors and its ligands [13, 34, 35]. Other nanoparticles with potential use in FCCS are fluorescent silica particles [36] and nanodiamonds with color centers [37, 38].
2.1.3 Fluorescent Proteins (FPs)
FPs are arguably the most widely used fluorophores for FCCS. They can be genetically encoded and thus guarantee specific labeling of the protein of interest with a defined stoichiometry. Although their photostability and brightness is inferior to organic dyes and QDs, and their complicated photophysics can make the interpretation of FCCS curves difficult (see Note 1), the ease of their usage and the fact that many protein-FP constructs are widely available, make them a good choice, as long as the relative concentration are considered (see Note 2). In addition, new FP variants with improved photophysical properties are continuously produced. For example, mKeima, a far-red fluorescent protein endowed with a large Stokes shift, is reported to be able to combine with both ECFP and EGFP for SW-FCCS [39]. Another FP with a large Stokes shift, LSSmOrange, is used to develop four-color single-laser FCCS solely based on FPs [40]. The most frequently used FP pair for DC-FCCS and SW-FCCS is mainly green and red pairs especially EGFP vs. mRFP/mCherry [10, 11, 21, 41], while TP-FCCS usually uses FPs with a large Stokes shift such as Cerulean and mCherry [42] or dyes with high two-photon absorption cross section such as Cyanine-5 (Cy5) and Rhodamine Green (RhGr) [43].
2.2 Instrumentation
FCS and FCCS instrumentation is commercially available from several manufacturers. Alternatively, commercial confocal microscopes can also be customized for the particular need of the user. The general FCCS setup and the correlation functions recorded are shown in Fig. 1, and more information about constructing FCCS systems can be found in literature [4, 6, 44]. The basic parameters for the parts of a standard system are as follows:
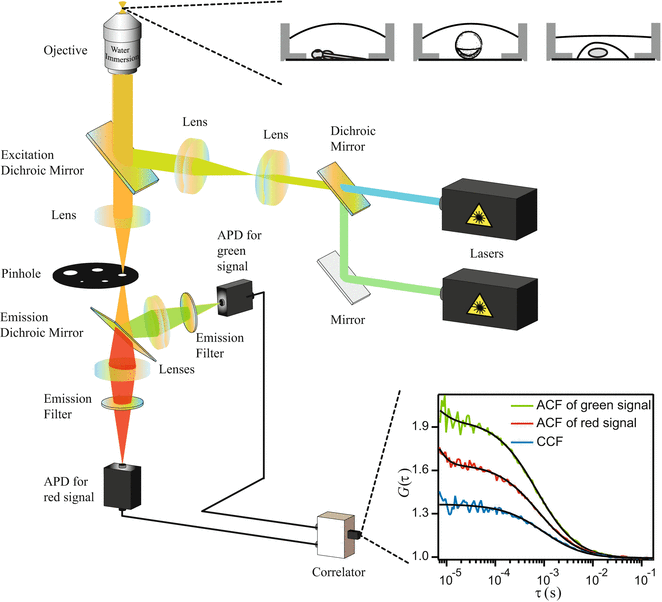
Fig. 1
General setup of an FCCS instrument and the illustration of ACF/CCF recorded
1.
Laser: To avoid photobleaching and saturation, which will alter auto- and cross-correlation functions (ACF/CCF), a laser power under 30 μW before entering the objective (in the case of an NA of 1.2) is recommended in most cases, and 5–30 μW are widely used [43, 45–48]. Use laser power as low as possible to reduce photobleaching but high enough to obtain good signal-to-noise ratio. The laser power should be constant for all experiments. A cps as low as 500 can be sufficient to generate useful CCFs [49], although typically cps of 1,000–5,000 is reached.
2.
Excitation/emission filters and the dichroic mirror: Filters should be chosen as a balance of maximizing the photons collected and minimizing cross talk. Filters with steep cut-ons/cutoffs are produced and help maximize light efficiency. The dichroic mirror should be selected to reduce cross talk as much as possible (see Note 3).
3.
Microscope objective: High-numerical aperture objectives with chromatic correction are recommended. For live biological samples, which are usually in an aqueous environment and in which deep penetration into tissue is required, water immersion objectives are more suitable than oil immersion objectives to avoid aberrations due to the refractive index mismatch between immersion and mounting media [50]. The objective correction collar, present on most microscope objectives used in FCCS, is designed to correct for the different thickness from different cover slides/glass bottom dishes. It must be adjusted every time before changing to another type of cover slips or chambers (see Subheading 3.2, step 4).
4.
Pinhole: The pinhole of a confocal microscope is used to constrain the detected light to the observation volume. A 60×, NA 1.2 water immersion objective with a pinhole size of 50 μm is practical to most biological samples and provides a focal volume of ~0.5–1 fL in size [11, 41] (see more in Subheading 3.3).
5.
Detector: Avalanche photodiodes (APDs) are highly sensitive photon counting devices and are widely used as the detectors for FCCS measurements. Photomultiplier tubes (PMTs), although they typically posses a lower quantum efficiency, can also be used.
6.
Correlator: In order to synchronize the signal from different channels and generate cross-correlations, both hardware and software correlators are widely used [17, 51–54]. While hardware correlators used to be preferred due to their better time resolution, the enhanced flexibility and low cost of software correlators and the continuing increase in speed and computational power of desktop computers make them a good alternative.
2.3 Choice of Sample
FCCS can be used for various samples, ranging from solutions to cell cultures and organisms. At the time of writing, live cells are probably the most widely used system for FCCS application, since cells provide a platform to mimic the natural environment for most biological molecules and are relatively easy to handle compared to organisms. That being said, FCCS has also been applied in organisms such as Drosophila and zebrafish [22, 55]. Penetration depth in living organisms can go as deep as 50–80 μm in FCS using one-photon excitation, or at least 200 μm with two-photon excitation, without adversely affecting the measured diffusion coefficient [22] (see Subheading 3.1).
3 Methods
3.1 Sample Preparation
3.1.1 Preparation of Dye Solutions for Calibration
1.
Stock solution: A defined amount of a solid organic dye is dissolved in DMSO or methanol to make a highly concentrated stock, typically in the mM range. It is stored at −20 °C or according to the instructions from the manufacturers. The dyes should not be exposed to light.
2.
Concentration determination: The stock can be diluted in deionized water or buffer solutions, depending on the dye and molar extinction coefficient, to the μM range so that the concentration can be confirmed using the molar extinction coefficient of the dye and the absorbance measurement in a spectrophotometer.
3.
Working solution: For FCCS measurement, the optimal concentration is in the 1–10 nM range [21, 56], corresponding to 0.3–6 molecules per focal volume (~0.5–1 fL). Working solutions can be kept at room temperature. Some organic dyes can easily attach to the plastic walls of their container. Hence shaking vigorously (or vortexing) before use is necessary. This can in some cases lead to deviations in measured concentrations when small volumes of dye solution are stored.
3.1.2 Cell Preparation
Recombinant fusion proteins for experiments with FPs can be transfected into cells by a variety of methods [57, 58], and protocols for cell transfections are provided by the relevant companies. In our experience, the different cell lines show different preferences to different methods and parameters being used, so the method and parameters have to be optimized for each case (see Note 2 for expression level control).
3.1.3 Embryo Specimen Preparation (Using Zebrafish as an Example)
Microinjection of RNA can be used to study proteins in early stage embryos, within a few hours postfertilization (hpf), while DNA microinjection is suitable for studies at a later stage, typically a few days postfertilization (dpf) [22].
The procedure is as follows: 2–10 pg of RNA/DNA is injected into a single cell or the yolk at an early stage, typically 1–32 cell stage. The embryos are incubated in egg water at 28.5 °C before being mounted in 0.5 % low-melting-temperature agar. The mounting procedure is performed under a stereoscope. To prevent pigmentation, PTU (0.003 % 1-phenyl-2-thiourea in 10 % Hank’s saline) is added at 18 hpf if a longer incubation period is needed. At stages after development of the central nervous system, onset of heartbeat and muscle contraction, the zebrafish might have to be anesthetized. This is accomplished by using, e.g., 0.01 % or ~500 μM tricaine [59, 60] (see Note 2 for expression level control).
3.2 Measurement Procedure
1.
System setup: Select the filters and dichroic mirrors according to the laser line(s) and fluorophores used. As overexposure of the APD to ambient room light can damage the APD, the room should be kept dark or make sure no ambient light can reach the APDs. In addition, ambient light can increase the background counts and influence the amplitudes of correlation functions. Count rates above 800 kHz should be avoided during measurements to prevent detector nonlinearities [4].
2.
Laser power determination: In typical confocal systems, the actual laser power is indicated as the percentage of the current. The dependence of the laser power on this percentage value is not linear, and the actual laser power has to be determined using a power meter. The laser power can be measured by removing the objective and placing the power meter in the laser’s path. Repeat the laser power measurement on a regular basis to confirm the stability of the lasers. Typically the laser power used is between 5 and 30 μW (see also Subheading 2.2). The cps values can vary for different systems and depend on the quality of the alignment, the filters used, and the overall transmission efficiency of the system. So they can vary even during a single measurement day, especially when the instrument room undergoes large temperature changes, and thus should be monitored. Atto 488 excited by a 5 μW 488 nm laser yields ~5–15 kHz with a 510 AF23 band-pass filter and 60×, NA 1.2 water immersion objective. Under the same experimental conditions, a cps of 3 kHz can be obtained from enhanced green fluorescent protein (EGFP) in live cells.
3.
Placement of the sample container: Type 1.5 (thickness 0.16–0.19 mm) and type 1.0 (thickness 0.13–0.16 mm) cover slides or glass bottom dishes are commonly used for the samples since they are usually within the range of the objective’s correction collar, 0.13–0.21 mm. Location of the sample by imaging is easy for a biological sample. However, for a solution sample or in cases where the position of the focal volume relative to the cover glass is important, the back reflection of the laser can be used to locate the position of the cover glass. This can be achieved by moving the laser focus from outside the cover glass into the sample while acquiring an image. The back reflection of the laser can be seen whenever the focal volume reaches the lower and upper side of the glass. Continue moving the objective at least 5 μm upward into the solution after seeing the upper side of the glass slide to avoid any distortion of the focal volume due to the glass slide.
4.
Calibration/pinhole adjustment: Before any measurement, calibration of the FCCS system is required for the optimal size of the focal volume as well as the optical efficiency of the system. The pinhole will reduce the signal-to-noise ratio if it is smaller than the optimal size, while the upper limits of a pinhole size can also be determined [61, 62] (see Subheading 3.3).
5.
Background determination: The background counts stem from four sources. The first source is ambient light from the room. This can be avoided by switching off all lights in the room. The second source is fluorescent impurities either from the buffer or from molecules in live cells (autofluorescence). They contribute about 300–500 Hz for buffer and 500–1,500 Hz for Chinese hamster ovary (CHO) cells for a laser power of 30 μW at 488 nm and fluorescence collected from 510 to 540 nm. Note that these values change with excitation wavelength and detection range but should be on the same order of magnitude. The third source is the detector, which possesses a certain amount of dark counts (20–500 counts per second, depending on the model). The dark counts, which are the signal the detector reports even in the total absence of light [63], are constant. In addition the detector produces additional counts due to afterpulsing, i.e., the recording of additional counts directly after actual photon detection events due to the detection method [63]. Afterpulsing has a finite probability to occur after each photon detection event, and thus this background is dependent on the actual counts detected. The last source, which is perhaps the least common, is due to the poor or wrong selection of dichroics and filters. This unintentionally results in back scattering of the laser, which is not filtered properly. This is easily detected by turning on the laser without any sample to check if there is any increase in the count rate. Background count rate should be determined for both channels before any experiments for data correction.
6.
Measurements: To obtain a precise result, concentrations of the fluorophores lower than 1 pM or higher than 100 nM are not recommended [64]. The count rate should range between 1 and 800 kHz. The measurement times depend on the signal-to-noise ratio of the particular measurement but typically range between 5 and 60 s. The measurement times in live cells should be kept as low as possible to avoid artifacts from cell movement and photobleaching but should be long enough to allow proper data analysis. Repeat the measurements 3–10 times for each position. Although there is no strict consensus on how long one can measure on a cell without inducing cell damage, to stay conservative we recommend measuring not more than three points in a cell where on each point one collects three consecutive measurements of 10–30 s with the settings as described here. For membrane measurements, ensuring that the membrane is in the center of the observation volume is important [65, 66]. There are two common ways to focus correctly at the upper membrane of a living cell. One is by observing the intensity trace: focus at the center of the nucleus first, and then while observing the intensity trace move the focus up until the strongest fluctuations, which stem from molecules in the plasma membrane, are observed. Another possibility is by using live imaging: focus at the center of the nucleus first, and then move the focus up until only the highest part of the upper membrane is in focus.
7.




Data analysis: Data analysis in FCS/FCCS is arguably the most difficult part, and new methods for the analysis of correlation data are being investigated to avoid data overinterpretation and helping in deciding between the numerous possible fitting models [67]. During fitting of the ACF and CCF, a few things have to be considered. First, the fitting range of the correlation functions has to be decided. The correlation function at times shorter than 1 μs is usually attributable to either the photodynamics of the fluorophore or the detector’s afterpulsing and is not fitted along. On the other end of the time scale of the correlation function, the function should have already decayed to 0 or 1 (depending on the correlation scheme), which is generally 0.1–1 s. There is no fixed rule for the exact range to fit the correlation function as samples, and fluorophore differs in each experiment. However, the same set of experiments should have a consistent fitting range. The second issue to note is the model used to fit the correlation function. In general, the ACFs are fitted using the simplest model consistent with the data, while the CCF is fitted with a single-diffusion component model without photodynamic contribution. Except in the case of strong cross talk, the photodynamics in the two channels are not correlated. Yet, in FCCS measurements often more than a single-diffusion time is found for a particular molecule. This might be because the molecules under investigation can be found in different states. For instance, a cytosolic fast diffusing molecule can bind to a membrane protein, or molecules in a lipid membrane might partition into different domains. However, if true interactions exist there must be at least one characteristic time, which is the same in all three correlation functions (two ACFs and the CCF). So the binding can only be confirmed by the increase in G CCF/G ACF (see next paragraph) compared to the negative control and similar diffusion times in the correlation functions [11].
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
