Fig. 1
FCCS setup. Light from two laser beams is focused into the sample using a highly corrected objective lens with a large numerical aperture. Fluorescence is guided via a pinhole into the detection unit where dichroic mirror and emission filters separate the light into two sensitive detectors. Diffusion of fluorescent molecules through the detection volume causes intensity fluctuations that can be analyzed by fitting the auto- and/or cross-correlation curves
Although not discussed in this chapter, alternative excitation strategies like TIRF, SPIM, STED, or spinning disk illumination can be combined with FCS, allowing the differently shaped and/or smaller observation volume. Furthermore, some of these options open the possibility to use FCS at higher dye concentrations [10–13].
A critical part of the equipment is the objective lens, especially when multicolor analysis is being used. It is essential to utilize an objective with a high numerical aperture (NA) in order to collect as many of the emitted photons as possible, during the limited residence time of a molecule in the detection volume. Most FCS data fitting models require a 3D Gaussian or Gaussian-Lorentzian-shaped observation volume, and therefore, any mismatch between the refractive index of the objective immersion liquid and the sample should be minimized. An objective with the best chromatic corrections is needed to have the best overlap between the multicolor excitation and detection volumes. Apochromat objectives fulfill these requirements and have a collar ring to manually correct for mismatches. For biological samples, a water-immersed lens is preferred since oil-immersed objectives, despite their higher NA values, lead to shape distortions of the observation volume and therefore result in poor data fits. Recently, a silicon-immersed objective with high NA was introduced by Olympus which approaches the refractive index of the cellular environment closely, resulting in high-quality intracellular FCCS curves [14].
However, even when using these highly corrected objective lenses, the Gaussian approximation of the detection volume is not perfectly met. If diffusion coefficients with extreme high accuracy should be obtained, a two-focus-FCS setup can be used where the cross-correlation signal of molecules diffusing between two closely spaced observation volumes, generated by a Wollaston prism in the excitation path, is analyzed [15].
For two-color analysis, a common problem is the presence of cross talk: the fluorescence of one type of dye detected in the “other” detector (e.g., the emission of the “green” dye in the “red” detector). The large tail in the dye emission spectrum causing this cross talk could give rise to false-positive cross-correlation. One could correct the data for this artifact, as discussed later in this chapter, but this issue can be prevented by using pulsed interleaved excitation (PIE) [16]. Most absorbance spectra are relatively narrow, and therefore, it is possible to excite each dye selectively using two specific laser lines. When two pulsed laser units are emitting in an alternating mode and at a frequency much faster than the residence time of the molecule in the observation volume, one can obtain the fluorescence of both dyes in different time windows. By time-gating the detected fluorescence, one can omit the cross-talk photons in the calculation of the auto- and cross-correlation curves.
2.2 Fluorescent Dyes
Since FCS is a method that relies on the analysis of relative fluorescence fluctuations, the average concentration of fluorescent molecules should be kept low, typically between 1 nM and 1 μM. During the passage of the molecules through the detection volume, as many photons as possible should be detected. As shown by Koppel [17], the molecular brightness of the fluorophore is one of the most important parameters determining the quality of an FCS measurement. In order to select a high-quality FCS dye from the multitude of dyes commercially available, the product of extinction coefficient times fluorescence quantum yield gives a good indication. Therefore, fluorescent beads or quantum dots will result in FCS curves with a high signal-to-noise ratio (SNR), but due to the relative large size of these beads and limited coupling possibilities, their application to biological samples is still limited. In addition, one has to consider other dye-related parameters as well like the sensitivity to photobleaching, environmental conditions that alter the dye characteristics (e.g., pH or hydrophobicity effects), the probability to go into a temporal dark state (triplet-state kinetics and blinking), and its stickiness to other molecules or to the sample holder.
2.2.1 Autofluorescence
For intracellular FCS measurements, the spectral profile of cellular autofluorescence should be taken into account. In many animal cells, the autofluorescence background is the strongest in the blue spectral region, while in plant cells, also a strong fluorescence is observed in the red region (>640 nm) when chlorophyll is present. Since (intracellular) FCS experiments are typically performed with nanomolar concentrations of the fluorescent probe, the autofluorescence of the cell could become a significant fraction of the total detected fluorescence. When the autofluorescence is not giving rise to an autocorrelation curve, a constant correction factor will be added to the data fitting model. It should be prevented that autofluorescence generates an autocorrelation curve since it is difficult to (quantitatively) correct for this contribution. It might be worth to put some effort in reducing the autofluorescence by optimizing growth conditions like varying the composition of the growth medium, changing to 37 °C and CO2 incubation of living animal cells, reducing the amount of light exposure in case of plant samples, or changing the experimental setup by shifting to another spectral detection region.
Common organic dyes used for FCS experiments are members of the Alexa (Invitrogen), Atto (ATTO-TEC), and CY (Amersham) families. When fluorescent proteins (FPs) are selected as the dye of choice (Table 1), one has to keep in mind that especially the red variants often show reduced maturation. Due to the delay time in forming a fluorescent chromophore, the true number of proteins will be underestimated. For example, when observing fusion proteins involved in yeast pheromone signaling, roughly 50 % of the mCherry fusion proteins were not visible [18]. This maturation issue is highly cell-type dependent, for example, in HeLa cells, only ~5 % of the mCherry fusions are not visible. Maturation ratios can be estimated by determining the concentration of green and red proteins in an FCS experiment using a < red FP-spacer protein-eGFP > fusion construct, assuming the eGFP protein to mature instantaneously. The large spacer protein present should prevent (FRET) between the two, which would complicate the analysis [9]. In order to increase the molecular brightness, multiple copies of the FPs could be introduced in the fusion protein, although distortion of the biological functionality and heterogeneous maturation should be prevented [18].
Table 1
Recommended standard monomeric fluorescent proteins for FC(C)S experiments
Fluorescent protein | λmax exc. (nm) | λmax em. (nm) | Comment | Reference |
|---|---|---|---|---|
TagBFP2 | 399 | 456 | Check maturation, UV excitation | [32] |
mTurquoise2 | 434 | 477 | very bright | [33] |
sGFP2 (or eGFP) | 495 | 512 | Photostable, bright | [34] |
sYFP2 (or mVenus) | 515 | 527 | Significant photophysics | [35] |
mCherry | 587 | 610 | Check maturation | [36] |
TagRFP-T | 555 | 584 | Photostable, bright, low maturation | [37] |
mKate2 | 588 | 635 | Check maturation | [38] |
2.2.2 Two-Color FCCS
For two-color FCCS studies, a critical point is the cross talk. Although one could correct for this artifact, it is desirable to diminish the spectral cross talk as much as possible. The popular FP pair CFP-YFP has a CFP bleed-through factor of 30 % in a typical setup. This will lead to significant false-positive cross-correlation, and therefore, corrections are required [18–20]. If the PIE excitation strategy is not possible, the quality of the measurements can be improved by choosing a pair that is spectrally more separated. However, when choosing such a pair, the overlap between the two detection volumes (which depends inversely on the spectral separation) will become less, and the maximum observable cross-correlation, and therefore the sensitivity, will decrease. Thus, a trade-off should be found between spectral separation and detection volume overlap. A good FCCS pair is eGFP-mCherry having high molecular brightness values, low photobleaching rates, and a reasonable spectral separation. Alternatively, one could use the more blue-shifted mTurquoise2-sYFP2 pair. When the fusion proteins give rise to FRET, the analysis is more complicated [21]. FRET will lower the molecular brightness of the donor molecule. Since the contribution of a molecule to the autocorrelation curve scales to the square of the molecular brightness [17], non-FRETting donor molecules that are brighter will dominate the correlation curve. Therefore, analysis can be simplified when a probe pair is chosen that will have a low FRET efficiency, e.g., due to a large spectral difference or spatial distance.
In order to minimize the importance of the correction factors and the uncertainty connected to it, the experimental conditions in two-color FCCS must be optimized. To lower the effect of cross talk, one should increase the signal of the red dye in the red detector. One could couple the red dye to the most abundant protein studied, lower the brightness of the green, and increase the brightness of the red dye by optimizing both laser powers or selecting a more red-shifted emission filter for the red detector [20]. To minimize the non-perfect overlap between the different observation volumes, dyes like the large Stokes shift fluorescent proteins [22] can be used. Here, multiple types of fluorescent proteins are excited using only a single laser line, while each FP fluoresces in a separate spectral window, due to their different Stokes shifts.
2.3 Data Analysis
Raw or correlated data can be fitted with dedicated analysis software. Many microscope manufacturers (e.g., Zeiss, ISS, Olympus, PicoQuant, and Becker & Hickl) distribute their own analysis software. In addition, commercial software packages can be purchased (e.g., SSTC FFS Data Processor and LFD SimFCS), and some user-written scripts for programs like Matlab, Igor, ImageJ, Origin, or Mathematica can be found on the Internet.
3 Methods
In the following section, a protocol is presented that can be used to measure dual-color fluorescence cross-correlation spectroscopy in living HeLa cells. Many issues discussed below will be similar in the case of single-color and/or in vitro FCS measurements. The idea behind this protocol is to study molecular interaction in the cell cytoplasm between sGFP2 and mCherry-labeled signaling proteins p110 and p85 [23] using an Olympus FV1000 microscope equipped with a PicoQuant PicoHarp detection unit. Although PIE excitation is possible with this setup, many FCS system do not have this feature and therefore is not employed in the protocols below.
3.1 Transfection of HeLa Cells
1.
Grow HeLa cells in DMEM medium (Gibco) supplemented with GlutaMAX (Gibco) in a live cell incubator at 37 °C with 5 % CO2.
2.
One day before transfection, transfer the cells from the culture flask to sterile circular (ø 24 mm, size 1) coverslips (Menzel-Gläser), stored in a six-well container (Greiner), at a confluency of approximately 60 % (~5.0 × 105 cells).
3.
Six hours after the cell transfer, replace the growth medium by phenol-free DMEM medium (Gibco), thereby lowering the autofluorescence.
4.
Transfect the cells using Lipofectamine in Opti-MEM according to the manufacturers protocol (Invitrogen). An important point to consider, especially when using high expression promoters in the construct(s), is that FCS requires low expression levels (see Note 1 ).
3.2 Microscope Calibration
1.
The microscope is placed in an air-conditioned room to ensure temperature stability and the calibration samples are stored in glass-bottomed 96-well plates (Whatman). In order to reduce the amount of detected background light, a protective blackened aluminum cover was placed on top of the sample during the measurements.
2.
Turn the microscope and lasers on roughly 30 min before starting the measurements in order to stabilize.
3.
Configure the FCS microscope filters: the sGFP2-labeled samples will be excited with the 488 nm laser line and the mCherry samples with the 561 nm diode laser (if available, use one or two pulsed lasers, suitable for the PIE excitation strategy, preventing cross talk). The fluorescence is separated from the excitation light by a dual-band dichroic filter, reflecting both the 488 and 561 nm excitation lines (Chroma). A secondary dichroic filter, LP562 (Chroma), separates the emission light into two different detection channels. Appropriate bandpass and longpass filters (e.g., BP500-550 (Semrock) for sGFP2 and LP572 (Chroma) for mCherry, respectively) are used for spectral selection and placed in front of the APD detectors (see Note 2 ).
4.
Measure the fluorescence of a ~100 nM solution of Alexa 488 (Invitrogen) in phosphate buffered saline (PBS) at a laser power of approximately 10–50 kW · cm−2. Adjust the correction collar of the objective to the position where the highest fluorescence count rate is observed in one of the two detection channels (see Note 3 ). In order to prevent that one optimizes scattered excitation light or adsorbed probe from the bottom of the sample holder, instead of observing the wished fluorescence signal, the focus should be set at least 20 μm into the solution.
5.
Optimize the detected fluorescence to its highest value by moving the X-, Y-, and, if available, Z-position of the pinhole(s) in the confocal microscope and optimize the lens position in front of the APD, if possible.
6.
Perform an FCS measurement of Alexa 488 and one of Atto 565 in PBS using their corresponding laser lines at 3.2 (488 nm) and 4.0 kW·cm−2 (561 nm) (see Note 4 ). Adjust the measurement time such that the resulting correlation curves are smooth in the decaying part of the curve.
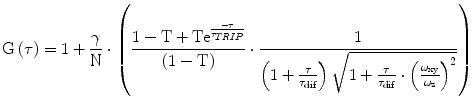
(1)
8.
Validate if the obtained fitting parameters are within the expected range. For our system, the value for a (=ω z /ω xy ) ranges from 4 to 15 and the values of τ dif for Alexa 488 or Atto 565 are around 35 and 60 μs, respectively. The observed brightness values vary between 8 and 12 kHz per molecule. Note that these values will be different for other microscope systems. In general, the higher the brightness per particle, the better the quality of the obtained data will be. The system has to be calibrated until the highest possible brightness is reached without having photobleaching and/or saturation artifacts [25].
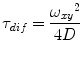
(2)
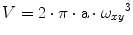
(3)
The effective cross-correlation observation volume is estimated from the cross-correlation curve, measuring a sample of purified sGFP2 in PBS (D = 90 μm2·s−1). Here we make use of the cross talk of sGFP2 since some of its emission will be detected in the mCherry detection channel. The emission of mCherry in the GFP channel can be omitted.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


