Familial Paraganglioma-Pheochromocytoma
Vania Nosé, MD, PhD
Key Facts
Terminology
Syndromes characterized by susceptibility to pheochromocytoma and paraganglioma resulting from germline mutations in SDHB, SDHC, and SDHD
Hereditary PGL/PCC should be considered in all individuals with multiple, bilateral, recurrent, or multifocal tumors
Clinical Issues
Germline mutations in SDHB are strongly associated with extraadrenal sympathetic paragangliomas
Mutations in SDHD and SDHC are associated with parasympathetic head and neck paragangliomas
Paragangliomas associated with germline SDHB mutation are more likely to become malignant
Up to 50% of persons with malignant extraadrenal paragangliomas have a germline SDHB mutation
Macroscopic Features
Fairly well-circumscribed, tan, rubbery-firm mass with fibrous pseudocapsule
Multifocal, bilateral, with multiple synchronous or metachronous tumors
Microscopic Pathology
Histological features of familial paragangliomas are similar to those of tumors that occur sporadically
Ancillary Tests
Immunohistochemical features are similar to those of sporadic tumors: Positive for neuroendocrine markers (chromogranin, CD56, and synaptophysin), negative for keratin
SDHB immunostain is decreased &/or absent in tumors of patients with SDHB or SDHD mutation
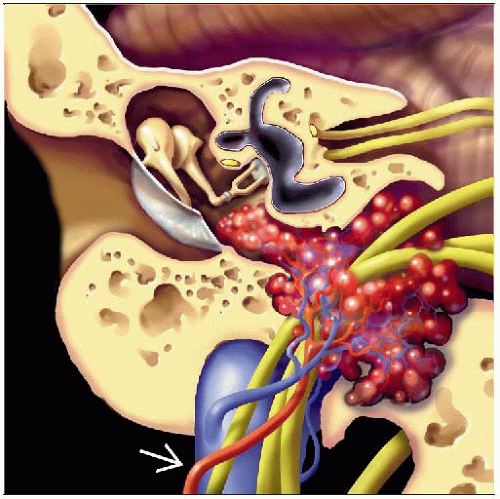 Coronal graphic shows glomus jugulare paraganglioma centered in the jugular foramen with superolateral extension into the middle ear. The ascending parapharyngeal artery  is feeding this vascular tumor. is feeding this vascular tumor. |
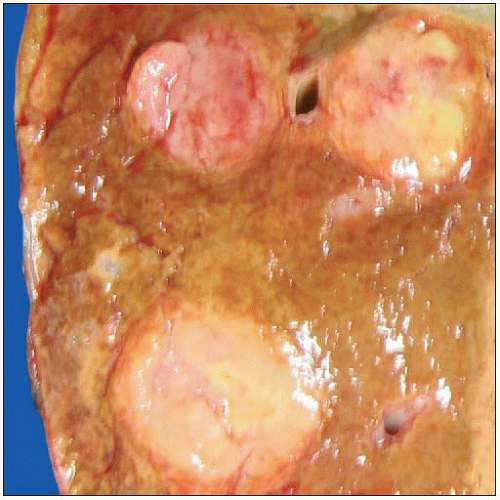 Gross cut surface of a liver from a patient with a hereditary malignant paraganglioma shows multiple well-circumscribed, firm, pale pink metastatic nodules. This patient also had metastases to the pancreas. |
TERMINOLOGY
Abbreviations
Hereditary paraganglioma-pheochromocytoma (PGL/PCC) syndromes
Synonyms
SDHA-, SDHB-, SDHC- and SDHD-related hereditary paraganglioma-pheochromocytoma syndromes
Definitions
Syndromes characterized by susceptibility to pheochromocytoma and paraganglioma resulting from germline mutations in SDHB, SDHC, SDHD, and newly described SDHA
Paragangliomas arise from neuroendocrine tissues symmetrically distributed along paravertebral axis from their predominant location at base of skull and neck to pelvis
Paragangliomas/pheochromocytomas are classified by location and secretory status
Sympathetic: Hypersecrete catecholamines
Parasympathetic: Do not hypersecrete catecholamines
Hereditary PGL/PCC syndromes should be considered in all individuals with PGL or PCC with the following findings
Multiple tumors, including bilateral tumors
Recurrent
Early onset (age < 40 years)
Multifocal with multiple synchronous or metachronous tumors
Family history of such tumors
Simplex cases: Many individuals with a hereditary PGL/PCC syndrome may present with solitary tumor of head or neck, thorax, abdomen, adrenal, or pelvis and no family history of the disorder
Paragangliomas in head and neck are primarily associated with parasympathetic nervous system and generally do not hypersecrete catecholamines or other hormones
Sympathetic paragangliomas located along paravertebral axis, and not in adrenal gland, are called extraadrenal sympathetic paragangliomas
Pheochromocytomas are catecholamine-secreting paragangliomas confined to adrenal medulla
Pheochromocytomas are also known as adrenal chromaffin tumors
Chromaffin cell/tumor is another term for any sympathetic (catecholamine-secreting) neuroendocrine cell/tumor regardless of location
Chromaffin refers to brown-black color that results from oxidization and polymerization of catecholamines contained in cells/tumors by chromium salts, such as potassium dichromate
Diagnosis of paragangliomas and pheochromocytomas is based on physical examination, imaging studies, biochemical testing, and pathology findings
CLINICAL ISSUES
Presentation
Symptoms of PGL/PCC result from either
Mass effect or
Catecholamine hypersecretion: Sustained/paroxysmal elevations in blood pressure, headache, episodic sweating, palpitations, pallor, and anxiety
Laboratory Tests
Catecholamines hypersecreted by PGL/PCC can be epinephrine (adrenaline), norepinephrine (noradrenaline), or dopamine
When a catecholamine-secreting tumor is suspected, plasma &/or 24-hour urinary fractionated metanephrine or catecholamines are evaluated for catecholamine hypersecretion
Measurement of fractionated metanephrine concentrations in plasma or urine is preferred
False-positive results may be reduced by follow-up testing for plasma chromogranin-A &/or urine fractionated metanephrine levels
Secretion of norepinephrine with little or no epinephrine suggests extraadrenal paraganglioma or pheochromocytoma associated with von Hippel-Lindau syndrome
Treatment
Treatment of manifestations
For secretory tumors including pheochromocytomas, antagonism of catecholamine excess followed by surgery
For nonsecretory head and neck paragangliomas, surgical resection
PGL/PCCs identified in SDHB-mutation-positive individuals require resection promptly because of high risk for malignant transformation
Genetic Counseling
Hereditary PGL/PCC syndromes are inherited in autosomal dominant manner
Mutations in SDHD (PGL1) demonstrate parent-of-origin effects and generally cause disease only when mutation is inherited from father
Each child of individual with hereditary PGL/PCC syndrome has 50% chance of inheriting diseasecausing mutation
Individual who inherits SDHD mutation from his/her mother has low but not negligible risk of developing disease
Individual who inherits SDHD mutation from his/her father is at high risk of manifesting paragangliomas and, to lesser extent, pheochromocytomas
Prenatal testing for pregnancies at increased risk is possible for families in which disease-causing mutation is known
Patient Evaluation
Includes detailed family history, including specific knowledge of any relatives with unexplained or incompletely explained sudden death
Personal medical history for the following
Symptoms of catecholamine excess: Sustained or paroxysmal elevations in blood pressure, headache, episodic profuse sweating, palpitations, pallor, and anxiety
Paroxysmal symptoms that may be triggered by changes in body position, increases in intraabdominal pressure, some medications, exercise, or micturition in the case of urinary bladder paragangliomas
Urinary bladder paragangliomas may also be accompanied by painless hematuria
Head and neck paragangliomas may present as enlarging masses that are asymptomatic or associated with symptoms of mass effects from size and location of tumors
Associated symptoms may include unilateral hearing loss, pulsatile tinnitus, cough, hoarseness of voice, pharyngeal fullness, swallowing difficulty, pain, and problems with tongue motion
Physical examination directed toward signs suggestive of PGL/PCC
Sympathetic paragangliomas and pheochromocytomas: Documentation of elevated blood pressure, tachyarrhythmias or other arrhythmias, and palpable abdominal masses
Genotype/Phenotype Correlation
Although persons with SDHB, SDHD, and SDHC mutations can develop pheochromocytomas or paragangliomas anywhere in paraganglia, the following correlations between gene involved and tumor location are used to guide diagnostic testing and in patient care
Germline mutations in SDHB are strongly associated with extraadrenal sympathetic paragangliomas
Chromaffin tumors in persons with germline SDHB mutations are 6x more likely to be extraadrenal than chromaffin tumors in general
Paragangliomas in persons with a germline SDHB mutation are more likely to become malignant than sporadic paragangliomas or in those persons with germline SDHD and SDHC mutations
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


