Evolution of pathogenic Escherichia coli
Sujay Chattopadhyay and Evgeni V. Sokurenko, University of Washington, Seattle, WA, USA
Introduction
As with evolution in general, bacterial evolution happens through the action of selection and drift on random genetic variations, affecting their frequency in nature, in space and in time. The evolution of virulence is viewed from the host’s perspective, where evolution of microbial genomes results in the ability of the microorganism to cause clinically manifested damage of the host. The ability to cause disease reflects the microbial fitness (i.e. ability to survive and reproduce) during the infection itself. To understand the driving forces and mechanisms behind the evolution of microbial virulence, one might compare the genomic content of organisms that are able to cause the disease to those that are unable to do so. There are relatively few (<200) bacterial species that are isolated as the cause of human infections, among which less than a dozen cause the vast majority of the infections. It is possible to compare these species to the species that do not act as common pathogens. While this approach is valid, the high level of genomic diversity between even closely related species makes such a strategy difficult. Frequent horizontal gene transfer between prokaryotic species results in a high level of genome mosaicism, which adds to the genome plasticity along with intragenomic variations, like point mutations, gene deletion/amplification, or genomic rearrangements. It is easier to compare organisms from the same bacterial species that differ significantly in their ability to cause disease. Escherichia coli offers an ideal example of such within-species virulence diversity.
Within-species diversity of pathogenic E. coli
There is a huge diversity of phenotypic traits across E. coli strains, both quantitatively or qualitatively, which allow these strains to differ in their appearance, behavior, metabolism, as well as in their ability to cause disease in humans. At one end, commensal strains, the vast majority of E. coli population, have adapted to colonize the host without causing disease. Colonization of pathogenic counterparts, on the other hand, can result in clinically significant pathologies. For example, the recommended regular dose of probiotic MutaFlor™, purported to promote health, contains billions of live bacteria of E. coli strain Nissle-1917, while the ingestion of only 100 bacteria of E. coli O157 strain or even fewer of Shigella flexneri can produce fatal disease (Todd et al., 2008; Allen et al., 2010). Also, the spectrum of disease caused by E. coli varies significantly, from diarrhea to meningitis, from asymptomatic bacteriuria to lethal urosepsis. This spectrum is caused by several main pathotypes of E. coli – enterotoxigenic (ETEC), enteropathogenic (EPEC), enterohemorrhagic (EHEC), enteroinvasive (EIEC/Shigella), enteroaggregative (EAEC) and extraintestinal-pathogenic (ExPEC, among which uropathogenic, UPEC, is the most common), as detailed elsewhere in this volume.
Despite being from the same species, different strains of E. coli are also highly diverse genetically. In fact, a given strain of E. coli shares only a minority of its genes with every other strain of the species (Rasko et al., 2008). This diversity is especially obvious from the analysis of clonally unrelated strains, i.e. those with different genotype profile based on the multi-locus sequence typing (MLST). MLST profile (or sequence type, ST) is defined in E. coli by comparing sequence identity of 400–500 bp long regions of seven housekeeping genes that are spread across the bacterial chromosome (Wirth et al., 2006).
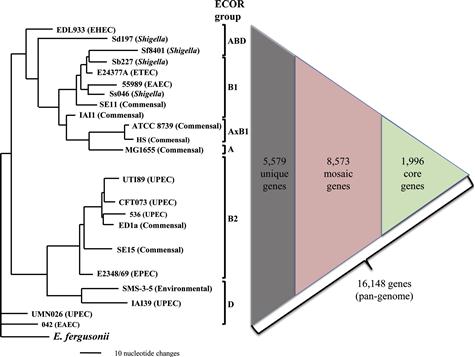
Here, to assess the level of clonal diversity of E. coli, we selected 22 clonally unrelated E. coli strains, ranging from commensal to different major pathotypes (Figure 3.1),with fully assembled annotated genome sequences publically available at the time of preparation of this chapter. The strains’ genome size ranged from 4116 to 5379 genes. However, based on 95% nucleotide identity and length preservation, a total of 16 148 genes were found in it least one of the 22 strains examined, comprising a minimal estimate of the E. coli pangenome. Out of these genes, 8573 genes were mosaic in nature, i.e. found in multiple (but not all!) strains that, on average, comprised 49–67% of individual genomes. Only 1996 genes were found in every strain and could be defined as core genes, which constituted from 37 to 49% of the genomes of individual strains. Finally, the rest of the genes either were found only in a single strain so far or were highly diverse orthologs (with less than 95% sequence identity) found in two or more strains.
The nucleotide-level difference found between the clonally unrelated E. coli was even greater. Even if only the shared genes more than 95% identical in sequence are considered, strain-to-strain polymorphisms affect over 100 000 nucleotides (2% of all gene sequences across the genome) on average. Out of these, about 20 000 nucleotide changes resulted in allelic amino acid replacements, with an average coded protein variant differing in 4–5 amino acids from the variant coded by the same gene but in a different strain.
Furthermore, strains from different pathotypes tend not only to be from different STs, but to belong to separate major phylogenetic clades that form so-called ECOR groups of E. coli (Figure 3.1) (Ochman and Selander, 1984). Thus, the genomic differences between strains from different pathotypes are even more pronounced than on average strain-to-strain, making the search for pathotype-specific traits difficult.
Interestingly, however, at least in some cases, clonally related strains with the same ST could be drastically different in their virulence (Weissman et al., 2012). For example, the probiotic strain Nissle-1917 that was isolated from the feces of a healthy individual almost a century ago has the same ST profile as model pathogenic E. coli strain CFT073 isolated from a patient with urosepsis in the 1980s (Vejborg et al., 2010). The vast majority of genes are shared between Nissle and CFT073, and only about 17% of genes are different. On the nucleotide level, there are about 100-fold fewer mutational differences than found between clonally unrelated strains. Thus, comparing clonally related strains might facilitate the search for the genetic basis of differential virulence and the evolutionary mechanisms of virulence acquisition.
Genetic mechanisms of virulence evolution
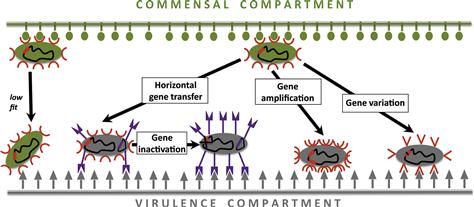
To gather insights on the microevolution of virulence in E. coli, the first step is to understand the genetic mechanisms the species adopt to achieve their within-species diversity. The major genetic events that underlie the evolution of virulence are: (a) horizontal transfer of novel genes, and (b) pathoadaptive mutation – amplification, inactivation, and variation – of existing genes (Figure 3.2).
Horizontal gene transfer (HGT)
This process, also called lateral gene transfer, involves transfer of genetic material from one strain to another, i.e. in contrast to vertical parent–progeny transfer of DNA during reproduction. According to a strict definition, HGT involves gene exchange between organisms from different species, but a more commonly used definition is applied to the gene transfer between organisms of the same species as well. HGT implies addition to the genome of novel genes, not an exchange by homologous recombination of different copies of the same gene, even though the mechanism of the gene movement between different strains could be the same.
HGT genomic elements
Genomic regions of HGT origin are typically carried by three major genetic elements in the E. coli: plasmids, prophages, and chromosomal islands.
Plasmids
Plasmids are usually circular and self-replicating molecules that co-exist with chromosomes. These extra-chromosomal elements harbor at least one, if not multiple, essential virulence determinants in all strains of diarrheagenic E. coli and Shigella spp. (Mellmann et al., 2009). One pioneering study demonstrated the pathogenic properties of plasmids in E. coli that causes diarrhea in piglets (Smith and Linggood, 1971). Subsequent studies showed that a major class of virulence factors encoded by the plasmids are genes conferring resistance to antimicrobial agents (e.g. cephalosporins, fluoroquinolones, aminoglycosides), toxic heavy metals (e.g. cadmium, mercury, silver), and other survival factors against lethal doses of antimicrobials (Mayer et al., 1995; Bennett, 2008; Hawkey, 2008). In addition, plasmid genes up-regulate important virulence and fitness genes in chromosomes through extensive cross-talk between plasmid and chromosome, as evidenced in many enteropathogenic E. coli (EPEC) strains (Schmidt, 2010). For example, the products of per genes in EPEC plasmids regulate expression of the pathogenicity locus of enterocyte effacement (LEE) genes (Mellies et al., 1999). Complex synergistic activities accelerate plasmid spread. For example, dense, structured populations as in biofilms increase the possibility of plasmid transfer by conjugation (see below) (Reisner et al., 2006). By the same token, the conjugation apparatus and the release of DNA stimulate formation and maintenance of biofilms (Molin and Tolker-Nielsen, 2003). Other examples of plasmid-borne virulence factors include Bundle-forming pili of EPEC (Donnenberg et al., 1992); EhxA in EHEC (Burland et al., 1998); pCoo in ETEC (Froehlich et al., 2005); plasmid-encoded toxin (Pet) in EAEC (Eslava et al., 1998); IcsA (VirG) in EIEC/Shigella (Buchrieser et al., 2000); TraT in UPEC (Timmis et al., 1985), etc.
Prophages
Temperate phages upon DNA injection into the host bacteria do not immediately enter into the lytic cycle but can instead integrate into the bacterial genome as a prophage. In fact, most of the mosaic gene elements in the E. coli genome are of a prophage nature (Canchaya et al., 2003). Most prophage genes are usually silent during bacterial growth and reproduction and are functional only when the prophage is activated (induced), i.e. enters the lytic pathway to produce active phages and lyse the host cell (usually under stress conditions, like UV light, antibiotic exposure, etc.), e.g. Shiga toxins Stx1 and Stx2 of EHEC (Dobrindt, 2005). However, some prophages commonly carry genes that are expressed and can add new phenotypic traits to their hosts that are important for success in colonization and competition within the habitat (Brussow et al., 2004). In EHEC and EPEC genomes, for example, a diversity of the prophages encode characteristic virulence factors (Ohnishi et al., 2002). A few examples are several type III secretion system (T3SS) effector proteins of EHEC and EPEC such as Cif (Marches et al., 2003), EspFU (Campellone et al., 2004), EspJ (Dahan et al., 2005), EspK (Vlisidou et al., 2006), NleA (Gruenheid et al., 2004), TccP (Garmendia et al., 2004) (see Chapters 4, 5, and 15). Other examples include cytolethal distending toxins in EPEC (Asakura et al., 2007); type II heat-labile enterotoxin in ETEC (Jobling and Holmes, 2012); GtrAB in EIEC/Shigella (Chaudhuri et al., 2010). Interestingly, until recently, there were no known major virulence determinants encoded by prophages in UPEC (Lavigne and Blanc-Potard, 2008).
Chromosomal islands
Chromosomal islands characterize a highly diverse group of DNA elements, with a broad range in size and abundance across the bacterial chromosomes (Dobrindt et al., 2004). Originally, actually, chromosomal islands were described as pathogenicity-associated islands (PAI), coined to define large unstable regions (10–200 kb) harboring virulence determinants on uropathogenic E. coli chromosomes and differing in GC content from the rest of the chromosome (Hacker et al., 1990). Examples of PAI-coded virulence factors are LEE island genes in EPEC and EHEC (McDaniel et al., 1995); Tia adhesin in ETEC (Fleckenstein et al., 2000); Protein involved in colonization (Pic) in EAEC (Henderson et al., 1999); SigA in EIEC/Shigella (Al-Hasani et al., 2000); Hemolysin in UPEC (O’Hanley et al., 1991). Besides pathogenicity-related functions, chromosomal islands encode genes representing a wide spectrum of traits such as mercury resistance and siderophore synthesis (Larbig et al., 2002), symbiosis (Sullivan et al., 2002), sucrose and aromatic compound metabolism (Gaillard et al., 2006), etc. The genomic islands are predicted using either sequence-based methods or comparative genomic analyses (Gal-Mor and Finlay, 2006). While sequence-based approaches rely on abnormal sequence composition (e.g. bias in GC content, dinucleotide frequency, codon usage) or on features associated with mobile genetic elements (e.g. presence of direct repeats, insertion sequence elements, tRNA, integrases, transposases, etc.), comparative analyses of multiple genomes are based on detection of genomic regions with anomalous phylogenetic patterns.
Mechanisms of HGT
There are three frequent genetic mechanisms that make the transfer of DNA happen: transduction, conjugation, and transformation. These diverse forms of transfer make HGT a very important process in expanding the potential of genetic adaptation in a bacterial species (Ochman et al., 2000).
Transduction
Transduction is defined as the movement of genetic material with the help of bacteriophages, the viruses that can inject DNA into the organisms. Lytic (virulent) phages that usually immediately multiply and lyze the infected bacterium, sometimes carry DNA from previously infected organisms, accidently packed into the capsule. Thus, if such DNA is injected instead of the viral genome, it can get incorporated in the infected microbial genome. However, while lytic phage transduction is commonly observed and utilized in lab experiments (e.g. P phage), it is unclear to what extent this process occurs in nature, because in these cases there are no traces of the viral DNA being tagged to the transduced DNA. Theoretically, any region of the bacterial genome could be transferred in this way, including plasmids and chromosomal islands and, thus, such a mechanism is termed ‘generalized transduction’ (Berg et al., 1983). It is a different story with the temperate phages that integrate into the bacterial chromosome as a prophage (see above). Its excision from the chromosome during the lytic cycle could be accompanied by accidental packaging (and then transfer) of non-phage DNA, but commonly these are the genomic regions flanking the prophage insertion site and, thus, this process is called ‘specialized transduction’ (Berg, et al., 1983). Still, any region of the genome could potentially be packaged in the activated prophage.
Conjugation
Conjugation involves direct transfer of DNA from donor to recipient microbial cell (Sorensen et al., 2005). E. coli and other Gram-negative bacteria have conjugation machinery that involves formation of a sex pilus, which is in contrast to the conjugation in Gram-positive bacteria that involves direct cell–cell contact adhesins (Grohmann et al., 2003). The pili and transfer function is encoded by conjugative plasmids that can be used as a vehicle by other (transmissible) plasmids or chromosomal regions (Zatyka and Thomas, 1998). Integration of the conjugative plasmid into the bacterial chromosome (e.g. integration of F plasmid episome into the chromosome via homologous recombination) can lead to the conjugative transfer of the flanking chromosomal regions (Holloway, 1993; Ambrozic et al., 1998), which could be, for example, chromosomal islands.
Virulence-associated genes can also reside in transposable elements that frequently change location between chromosome and plasmid and, in this way, can move horizontally. These elements can be insertion sequences (IS elements), i.e. small mobile fragments with short terminal inverted repeat sequences. These entities can also be transposons, with or without flanking insertion sequences termed composite and non-composite transposons respectively. Many transposons include antibiotic-resistance genes, e.g. Tn5 carrying kanamycin, bleomycin, and streptomycin resistance genes (Reznikoff, 2008). Also, integrons represent multiple classes of genetic elements triggering the spread and acquisition of gene cassettes by conjugation (Mazel, 2006). They specify an integrase, attachment sites, and transcriptional elements that ensure proper expression of exogenous antibiotic-resistance determinants in recipient genomes. Examples are MDR efflux pumps, carbapenemases, etc. (Giedraitiene et al., 2011).
Transformation
Transformation is the direct uptake of naked DNA, often followed by recombination in the genomic DNA of recipient strain. This HGT process incorporates large discrete DNA segments as chromosomal islands (Chen et al., 2005). While the phenomenon was known and used for decades in experiments with laboratory strains of E. coli, natural transformation of wild-type E. coli strains has been described relatively recently (Baur et al., 1996). Potentially any type of DNA region could be transferred in this way. Another mechanism that is somewhat similar to the transformation, which does not involve a specialized genetic vehicle but where the DNA is not floating ‘naked’, is outer membrane vesicle-mediated transfer in E. coli (Yaron et al., 2000). However, unlike the transduction and conjugation, transformation mechanisms are not well understood in E. coli.
Pathoadaptive mutations
Pathoadaptive mutation is an alternative mechanism for the virulence evolution that involves genetic alteration of the existing genome components rather than acquisition of novel genes. The major mechanisms are gene amplification, inactivation, and variation (Figure 3.2).
Gene amplification
Amplification of gene copies can lead to increased production of the corresponding protein and, if that confers an advantage for the pathogen, be selected during the infection. It has been proposed recently that transient amplification of genetic regions is a common phenomenon in E. coli (and other bacteria), allowing for fast adaptation in environments and increased rates of adaptive evolution (Kugelberg et al., 2006). Thus, gene amplification might be a more significant phenomenon than appreciated previously. Examples of virulence factors with multiple copies are Acriflavin resistance protein D (AcrD) in EPEC (Fukiya et al., 2004); Iha adhesin in EHEC (Mellmann et al., 2009); P-fimbrial genes in some UPEC (Kao et al., 1997).
Gene duplication is another critical event commonly found in many enteric pathogens including E. coli (Ohno, 1970; Himmelreich et al., 1996; Tomb et al., 1997), providing means for novel functional evolution. An important example is the duplication of Shiga toxin 2 encoding genes (stx2) found in some EHEC O157:H7 outbreak strains (Muniesa et al., 2003), and in vitro experiments showed correlation of the occurrence of duplicated genes with increased production of Stx2 (Bielaszewska et al., 2006). Studies showed that evolution of orthologs faces considerable selective constraints (Hughes and Hughes, 1993), and the level of functional constraints reduces immediately following a duplication event resulting in a significantly higher rate of evolution between paralogs (Lynch and Conery, 2000; Jordan et al., 2002; Kondrashov et al., 2002).Thus, duplication allows the emergence of proteins with novel structure and functions. For example, the cspA (the major cold-shock protein) family homologs in E. coli resulted from a number of gene duplication events allowing emergence of specific groups of genes that adaptively respond to different environmental stresses such as cold-shock stress (cspA, cspB, cspG), nutritional deprivation (cspD), etc. (Yamanaka et al., 1998).
Gene inactivation (anti-virulence factors)
HGT and gene amplification are so-called the ‘gain-of-function’ mechanisms for the evolution of microbial virulence. Another and quite common mechanism represents ‘loss-of-function’ that includes gene or coded protein inactivation via point substitution, frameshift mutation, insertion sequence acquisition, and gene deletion. This mechanism is often crucial to enhance the fitness of a pathogen in a novel environment.
It is necessary to distinguish between gene loss due to pathoadaption and due to reductive evolution. The model of reductive evolution is based on the lack of universal need of some genes across different niches. So, genes that are essential in one habitat but are not required in a novel habitat could be lost based on the use-it-or-lose-it principle, especially if the habitat shift is permanent. The latter trajectory of evolution develops when certain bacteria change their lifestyle to an obligate intracellular pathogen, and permanently shed off genes non-essential within the host (Moran and Plague, 2004). Mycobacterium leprae (Cole et al., 2001), Coxiella burnetii (Seshadri et al., 2003), the Rickettsiae (Andersson et al., 1998) are some examples of reductive genome evolution. Moreover, the restricted intracellular lifestyle within a host reduces the possibility of HGT events thereby maintaining a reduced genome size (Moran and Plague, 2004). However, loss of gene by such a mechanism is not driven by positive selection pressures for it, and therefore is not adaptive per se. The unnecessary genes are either not expressed or their expression does not provide fitness advantage or significant disadvantage. Thus, their loss rather happens due to the lack of negative selection against it and is driven by genetic drift and not positive selection.
Alternatively, loss of certain genes could be highly adaptive, especially for pathogens. Indeed, genes that are essential for the fitness in one habitat might be not only useless but also detrimental for the fitness in another habitat (Maurelli, 2007). This will force the emergence of new variants of the bacterium through elimination of genes that, for example, down-regulate those genes, increased expression of which is important for the fitness in the new niche. Another example would be elimination of genes, the product of which either directly inhibits the function of critical traits or provides liability in the new habitat, e.g. those coding for the surface structure recognized well by innate or adaptive immune mechanisms.
Genes, the function of which is detrimental to virulence, are sometimes termed ‘antivirulence factors’. A great example of an antivirulence gene is cadA in Shigella spp. The gene cadA encodes lysine decarboxylase (LDC) and is expressed in >90% of E. coli isolates (Maurelli, 2007). However, all strains of Shigella as well as the enteroinvasive E. coli (EIEC that cause dysentery similar to shigellosis, see Chapter 7) lack expression of LDC (Silva et al., 1980). Experiments in an animal model showed that if Shigella was forced to produce LDC via transformed cadA, it remained invasive but its enterotoxic activity was significantly diminished compared to the wild-type level (Maurelli et al., 1998). The product of lysine decarboxylation is cadaverine, which inhibits the function of plasmid-encoded enterotoxins in Shigella. Another experiment showed the role of cadaverine in preventing the ability of Shigella to stimulate transepithelial migration of polymorphonuclear neutrophils (McCormick et al., 1999). Since expression of LDC followed by production of cadaverine attenuates virulence phenotypes, cadA in Shigella satisfies the necessary features of an antivirulence gene (Maurelli, 2007). Another example of an antivirulence gene, again in Shigella, is ompT, a prophage-mediated gene encoding a surface protease. This protease degrades IcsA/VirG, a virulence protein in Shigella that is required for keratoconjunctivitis in guinea pigs and is involved in intra- and intercellular motility (Nakata et al., 1993). The gene ompT is lacking in all lineages of Shigella, also fulfilling the criteria to be an antivirulence gene. Multiple independent evolutionary origins of Shigella strains indicate convergent evolution via the loss of antivirulence factors, including inactivation/deletion of LDC by different mechanisms, which is a strong indication for the action of positive selection (Pupo et al., 2000).
Even a clean single gene deletion, however, could potentially result in toxic effects by disrupting, for example, biogenesis pathways and build-up of intermediate components (Holt et al., 2008). That is possibly why multiple gene or entire gene operons/clusters tend to be lost, resulting in so-called ‘black holes’ in the genomes (Maurelli, et al., 1998). Instead of gene deletion, ‘loss of function’ can also be achieved by a mutation introducing premature stop codons or by frameshift mutations that disrupt the reading frame. However, such mechanisms result in expression of truncated and/or misfolded proteins that could be toxic for the bacterial cell (Kuo and Ochman, 2010). In contrast, loss-of-function due to mutation in the active site of the protein might provide a better way to achieve the adaptive effect. Mutations in the active protein site have been reported, for example, in the major, type 1 fimbrial adhesin of E. coli species, FimH, where inactivating mutations are found in the mannose-binding pocket of the adhesive protein of some EHEC isolates (Shaikh et al., 2007). Loss of the type 1 fimbrial function has been also seen for Shigella and is likely to provide some kind of pathogenicity-adaptive effect in enteric pathogens (Snellings et al., 1997).
Gene variation
Point mutations that do not inactivate but modify the function of coded protein are another important player of the pathoadaptive mechanism of E. coli evolution. An example of pathoadaptive point mutation is the evolution of fimH gene encoding the type 1 fimbrial adhesin in E. coli (see Chapter 12). FimH is expressed by >90% of E. coli (Johnson and Stell, 2000), and uropathogenic isolates express some specific variants of FimH owing to accumulation of point mutations (Weissman et al., 2006). These variants significantly enhance binding to mannosylated glycoproteins on uroepithelial cell surfaces, thereby increasing bacterial tropism to uroepithelium (Sokurenko et al., 1998; Hommais et al., 2003). Point mutations leading to functional modification that is pathoadaptive in nature have been shown in at least two other types of E. coli adhesins – Dr family (Korotkova et al., 2007) and class 5 fimbrial adhesins in enterotoxigenic isolates (Tchesnokova et al., 2010; Chattopadhyay et al., 2012).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


