Chapter 4 Evaluation of drugs in humans
Experimental therapeutics
Modern medicine is sometimes accused of callous application of science to human problems and of subordinating the interest of the individual to those of the group (society).1 Official regulatory bodies rightly require scientific evaluation of drugs. Drug developers need to satisfy the official regulators and they also seek to persuade the medical profession to prescribe their products. Patients, too, are more aware of the comparative advantages and limitations of their medicines than they used to be. To some extent, this helps encourage patients to participate in trials so that future patients can benefit, as they do now, from the knowledge gained from such trials. An ethical framework is required to ensure that the interests of the individual participant take precedence over those of society (and, more obviously, those of an individual or corporate investigator).
Research involving human subjects
The definition of research continues to present difficulties. The distinction between medical research and innovative medical practice derives from the intent. In medical practice the sole intention is to benefit the individual patient consulting the clinician, not to gain knowledge of general benefit, though such knowledge may incidentally emerge from the clinical experience gained. In medical research the primary intention is to advance knowledge so that patients in general may benefit; the individual patient may or may not benefit directly.2
Consider also the process of audit, which is used extensively to assess performance, e.g. by individual health-care workers, by departments within hospitals or between hospitals. Audit is a systematic examination designed to determine the degree to which an action or set of actions achieves predetermined standards. Whereas research seeks to address ‘known unknowns’ and often discovers ‘unknown unknowns,3 audit is limited to the monitoring of ‘known knowns’: maybe important, but clearly limited.
Ethics of research in humans4
Some dislike the word ‘experiment’ in relation to humans, thinking that its mere use implies a degree of impropriety in what is done. It is better that all should recognise from the true meaning of the word, ‘to ascertain or establish by trial’,5 that the benefits of modern medicine derive almost wholly from experimentation and that some risk is inseparable from much medical advance.
The moral obligation of all doctors lies in ensuring that in their desire to help patients (the ethical principle of beneficence) they should never allow themselves to put the individual who has sought their aid at any disadvantage (the ethical principle of non-maleficence) for ‘the scientist or physician has no right to choose martyrs for society’.6
In principle, it may be thought proper to perform a therapeutic trial only when doctors (and patients) have genuine uncertainty as to which treatment is best.7 Not all trials are comparisons of different treatments. Some, especially early phase trials of new drugs, are comparisons of different doses. Comparisons of new with old should usually offer patients the chance of receiving current best treatment with one which might be better. Since this is often rather more than is offered in resource-constrained routine care, the obligatory patient information sheet mantra that ‘the decision whether to take part has no bearing on your usual care’ may be economical with the truth. But it is also simplistic to view the main purpose of all trials with medicines as comparative.
The ethics of the randomised and placebo-controlled trial
Providing that ethical surveillance is rooted in the ethical principles of justice,8 there should be no difficulty in clinical research adapting to current needs. And even if the nature of early phase research is changing, the randomised controlled trial will remain the cornerstone of how cause and effect is proven in clinical practice, and how drugs demonstrate the required degree of efficacy and safety to obtain a licence for their prescription.
The use of a placebo (or dummy) raises both ethical and scientific issues (see placebo medicines and the placebo effect, Ch. 2). There are clear-cut cases when placebo use would be ethically unacceptable and scientifically unnecessary, e.g. drug trials in epilepsy and tuberculosis, when the control groups comprise patients receiving the best available therapy.
The pharmacologically inert (placebo) treatment arm of a trial is useful:
• To distinguish the pharmacodynamic effects of a drug from the psychological effects of the act of medication and the circumstances surrounding it, e.g. increased interest by the doctor, more frequent visits, for these latter may have their placebo effect. Placebo responses have been reported in 30–50% of patients with depression and in 30–80% with chronic stable angina pectoris.
• To distinguish drug effects from natural fluctuations in disease that occur with time, e.g. with asthma or hay fever, and other external factors, provided active treatment, if any, can be ethically withheld. This is also called the ‘assay sensitivity’ of the trial.
• To avoid false conclusions. The use of placebos is valuable in Phase 1 healthy volunteer studies of novel drugs to help determine whether minor but frequently reported adverse events are drug related or not. Although a placebo treatment can pose ethical problems, it is often preferable to the continued use of treatments of unproven efficacy or safety. The ethical dilemma of subjects suffering as a result of receiving a placebo (or ineffective drug) can be overcome by designing clinical trials that provide mechanisms to allow them to be withdrawn (‘escape’) when defined criteria are reached, e.g. blood pressure above levels that represent treatment failure. Similarly, placebo (or new drug) can be added against a background of established therapy; this is called the ‘add on’ design.
• To provide a result using fewer research subjects. The difference in response when a test drug is compared with a placebo is likely to be greater than that when a test drug is compared with the best current, i.e. active, therapy (see later).
• The severity of the disease.
• The effectiveness of standard therapy.
• Whether the novel drug under test aims to give only symptomatic relief, or has the potential to prevent or slow up an irreversible event, e.g. stroke or myocardial infarction.
• The objective of the trial (equivalence, superiority or non-inferiority; see p. 45). Thus it may be quite ethical to compare a novel analgesic against placebo for 2 weeks in the treatment of osteoarthritis of the hip (with escape analgesics available). It would not be ethical to use a placebo alone as comparator in a 6-month trial of a novel drug in active rheumatoid arthritis, even with escape analgesia.
The precise use of the placebo will depend on the study design, e.g. whether crossover, when all patients receive placebo at some point in the trial, or parallel group, when only one cohort receives placebo. Generally, patients easily understand the concept of distinguishing between the imagined effects of treatment and those due to a direct action on the body. Provided research subjects are properly informed and give consent freely, they are not the subject of deception in any ethical sense; but a patient given a placebo in the absence of consent is deceived and research ethics committees will, rightly, decline to agree to this. (See also: Lewis et al (2002) in Guide to further reading, at the end of this chapter.)
Injury to research subjects9
The question of compensation for accidental (physical) injury due to participation in research is a vexed one. Plainly there are substantial differences between the position of healthy volunteers (whether or not they are paid) and that of patients who may benefit and, in some cases, who may be prepared to accept even serious risk for the chance of gain. There is no simple answer. But the topic must always be addressed in any research carrying risk, including the risk of withholding known effective treatment. The CIOMS/WHO Guidelines4 state:
Payment of subjects in clinical trials
There is an intuitive abreaction by physicians to pay patients (compared with healthy volunteers), because they feel the accusation of inducement or persuasion could be levelled at them, and because they assuage any feeling of taking advantage of the doctor–patient relationship by the hope that the medicines under test may be of benefit to the individual. This is not an entirely comfortable position.10
Rational introduction of a new drug to humans
Phases of clinical development
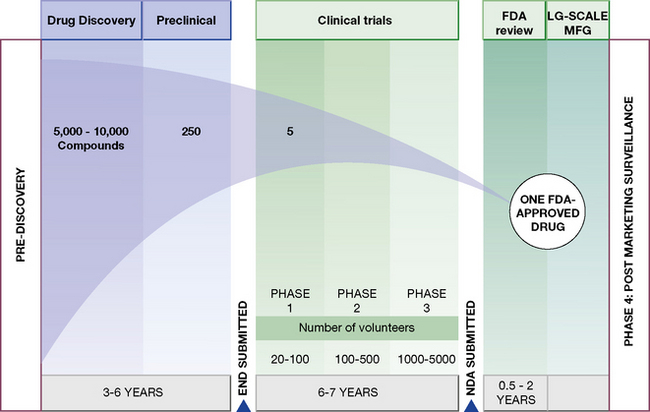
Human experiments progress in a commonsense manner that is conventionally divided into four phases (Fig. 4.1). These phases are divisions of convenience in what is a continuous expanding process. It begins with a small number of subjects (healthy subjects and volunteer patients) closely observed in laboratory settings, and proceeds through hundreds of patients, to thousands before the drug is agreed to be a medicine by a national or international regulatory authority. It is then licensed for general prescribing (though this is by no means the end of the evaluation). The process may be abandoned at any stage for a variety of reasons, including poor tolerability or safety, inadequate efficacy and commercial pressures. The phases are:
• Phase 1. Human pharmacology (20–50 subjects):



• Phase 2. Therapeutic exploration (50–300 subjects):

 pharmacokinetics and pharmacodynamic dose ranging, in carefully controlled studies for efficacy and safety,11 which may involve comparison with placebo.
pharmacokinetics and pharmacodynamic dose ranging, in carefully controlled studies for efficacy and safety,11 which may involve comparison with placebo.• Phase 3. Therapeutic confirmation (randomised controlled trials; 250–1000 + subjects):


• Phase 4. Therapeutic use (pharmacovigilance, post-licensing studies) (2000–10 000 + subjects):

Official regulatory guidelines and requirements12
For studies in humans (see also Ch. 6) these ordinarily include:
• Studies of pharmacokinetics and bioavailability and, in the case of generics, bioequivalence (equal bioavailability) with respect to the reference product.
• Therapeutic trials (reported in detail) that substantiate the safety and efficacy of the drug under likely conditions of use, e.g. a drug for long-term use in a common condition will require a total of at least 1000 patients (preferably more), depending on the therapeutic class, of which (for chronic diseases) at least 100 have been treated continuously for about 1 year.
• Special groups. If the drug will be used in, for example, the elderly or children, then these populations should be studied. New drugs are not normally studied in pregnant women. Studies in patients having disease that affects drug metabolism and elimination may be needed, such as patients with impaired liver or kidney function.
• Fixed-dose combination products will require explicit justification for each component.
• Interaction studies with other drugs likely to be taken simultaneously. Plainly, all possible combinations cannot be evaluated; a rational choice, based on knowledge of pharmacodynamics and pharmacokinetics, is made.
• The application for a licence for general use (marketing application) should include a draft Summary of Product Characteristics for prescribers. A Patient Information Leaflet must be submitted. These should include information on the form of the product (e.g. tablet, capsule, sustained-release, liquid), its uses, dosage (adults, children, elderly where appropriate), contraindications, warnings and precautions (less strong), side-effects/adverse reactions, overdose and how to treat it.
The emerging discipline of pharmacogenomics seeks to identify patients who will respond beneficially or adversely to a new drug by defining certain genotypic profiles. Individualised dosing regimens may be evolved as a result (see p. 101). This tailoring of drugs to individuals is consuming huge resources from drug developers but has yet to establish a place in routine drug development.
Therapeutic investigations
• The therapeutic effect itself (sleep, eradication of infection), i.e. the outcome.
• A surrogate effect, a short-term effect that can be reliably correlated with long-term therapeutic benefit, e.g. blood lipids or glucose or blood pressure. A surrogate endpoint might also be a pharmacokinetic parameter, if it is indicative of the therapeutic effect, e.g. plasma concentration of an antiepileptic drug.
Therapeutic evaluation
The aims of therapeutic evaluation are three-fold:
1. To assess the efficacy, safety and quality of new drugs to meet unmet clinical needs.
2. To expand the indications for the use of current drugs (or generic drugs13) in clinical and marketing terms.
3. To protect public health over the lifetime of a given drug.
The process of therapeutic evaluation may be divided into pre- and post-registration phases (Table 4.1), the purposes of which are set out below.

If the drug is found useful in these trials, it becomes desirable next to find out how closely the ideal may be approached in the rough and tumble of routine medical practice: in patients of all ages, at all stages of disease, with complications, taking other drugs and relatively unsupervised. Interest continues in all patients from the moment they are entered into the trial and it is maintained if they fail to complete, or even to start, the treatment; the need is to know the outcome in all patients deemed suitable for therapy, not only in those who successfully complete therapy.14
Need for statistics
In order truly to know whether patients treated in one way are benefited more than those treated in another, it is essential to use numbers. Statistics has been defined as ‘a body of methods for making wise decisions in the face of uncertainty’.15 Used properly, they are tools of great value for promoting efficient therapy. More than 100 years ago Francis Galton saw this clearly:
The human mind is … a most imperfect apparatus for the elaboration of general ideas … In our general impressions far too great weight is attached to what is marvellous … Experience warns us against it, and the scientific man takes care to base his conclusions upon actual numbers … to devise tests by which the value of beliefs may be ascertained.16
Concepts and terms



