CHAPTER 23 Essential thrombocythemia and primary myelofibrosis
Introduction
Among the classical Philadelphia-chromosome-negative myeloproliferative neoplasms (MPN), essential thrombocythemia (ET) and primary myelofibrosis (PMF) represent two subtypes with considerable overlap in clinical and hematological presentation in particular at early stages of disease.1 Differentiation between those two MPN subtypes is of clinical relevance, since PMF is characterized by a considerably higher risk of progression and leukemic transformation. In contrast, ET generally represents a stable disease with only minimal risk of myelofibrotic progression.2 There is also a significant difference in long-term survival between both groups. The 15-year age-adjusted relative survival rate for ET is nearly 84%. In contrast, PMF is characterized by a significant loss in life expectancy. Even when diagnosed in very early stages of disease, PMF patients have a 15-year survival of only 55–67%.2 Although the discovery of the JAK2V617F mutation (see Chapter 21) led to important progress in the understanding of the molecular pathogenesis of ET and PMF,3 the finding of this mutation in several MPN subtypes and the absence of the JAK2V617F allele in many MPN patients preclude the use of JAK2V617F testing alone to establish a diagnosis of ET or PMF. This is reflected in the revised WHO diagnostic criteria for ET and PMF (Box 23.1), which include mutation screening in the diagnostic work-up, but explicitly require a bone marrow (BM) morphological examination for making the diagnosis of both ET and PMF. Although a significantly lower JAK2V617F allele burden has been found in ET when compared to early stages of PMF,4 such variability in the allele burden does not represent a sufficient criterion for distinguishing among different clinical entities. Furthermore, JAK2V617F-negative cases reveal a similar clinical profile and outcome5 and increase in JAK2V617F allele frequencies is not linked to myelofibrotic transformation or progression to acute myeloid leukemia (AML).6
Box 23.1 WHO criteria for the diagnosis of essential thrombocythemia (ET) and primary myelofibrosis (PMF)
* In prefibrotic/early stages these features are generally only borderline expressed.
a) Causes of reactive thrombocytosis include iron deficiency, splenectomy, surgery, infection, inflammation, connective tissue disease, metastatic cancer, and lymphoproliferative disorders. The presence of a condition associated with reactive thrombocytosis does not exclude the possibility of ET if the first three criteria are met.
b) Causes of bone marrow fibrosis include infection, autoimmune disorders or other chronic inflammatory conditions, hairy cell leukemia or other lymphoid neoplasm, metastatic malignancy, or toxic (chronic) myelopathies.
ET and PMF usually affect the elderly population, but they can occasionally be found in children, and in this instance, they raise age-related diagnostic and management issues.7 Familial clustering of ET and also PMF is known, and this observation led to a suggestion of predisposition alleles even before the discovery of the JAK2V617F mutation.8 Relatives of patients with ET have a more than sevenfold increase in the relative risk of developing a similar MPN.9 However, the coexistence of different clinical entities and of JAK2V617F-positive and JAK2V617F-negative diseases in the same family is noteworthy.8,10
When addressing the current WHO criteria (Box 23.1) for diagnosing the different MPN entities,11,12 it is essential to emphasize that these guidelines do not claim that a single histological parameter defines a subgroup, but that the different subtypes of MPN are characterized by specific morphological BM patterns.13 These patterns are composed of distinctive features and should always be reviewed in close relation to clinical, hematological and molecular-genetic findings to achieve a consensus-based working diagnosis.1,3,14,15 In contrast to the determination of age-dependent cellularity and to the semiquantitative grading of myelofibrosis,16,17 characterization of megakaryopoiesis may cause significant difficulties concerning definition and easy recognition of disease-related patterns among different observers.18 Assessment of megakaryocytic histotopography, i.e. their arrangement within the BM space, and detection of certain nuclear abnormalities (besides maturation defects) are the keys to the diagnosis.19 In contrast to the normal BM, in which megakaryocytes show a central distribution of single isolated cells, the overall increase of megakaryocytes in MPN is often associated with the uneven distribution such as group formation: from small clusters (at least three cells) to extensive groups (more than seven cells).20 These megakaryocyte clusters may display either a loose (intermingled with other hematopoietic cells) or dense arrangement.19 An abnormal dislocation of megakaryocytes towards the endosteal (paratrabecular) border is a highly conspicuous finding that is usually not seen in reactive disorders. Other features indicating a neoplastic process are peculiar nuclear aberrations and maturation defects that imply disturbances of the normal development of megakaryopoiesis.1,21–23 These include:
Furthermore, maturation defects include a conspicuous deviation of the nuclear-cytoplasmic ratio or maturation with appearance of bizarre megakaryocytes. It is noteworthy that all these changes may be present in megakaryocytes of different sizes and ploidy status. Finally, so-called naked (denuded, bare) nuclei with condensed chromatin pattern may frequently be found implicating a stimulated thrombocyte shedding and cell turnover.20
In other hematopoietic lineages, an increase and left-shifting of neutrophil granulopoiesis or erythropoiesis may be a prominent feature or a reduction in the amount of nucleated red cell precursors may be found, depending on disease entity and phase.24
Essential thrombocythemia (ET)
Essential thrombocythemia (ET) is a MPN that involves primarily the megakaryocytic lineage. It is characterized by sustained thrombocytosis >450 × 109/l in the peripheral blood (PB), increased numbers of large, mature megakaryocytes in the BM, and clinically by episodes of thrombosis and/or hemorrhage. Because there is no known genetic or biological marker specific for ET, other causes for thrombocytosis such as other MPN, inflammatory and infectious disorders, hemorrhage and other types of hematopoietic and non-hematopoietic neoplasms must be excluded.12 The presence of the BCR-ABL1 fusion gene excludes the diagnosis of ET (Box 23.1). The reported annual incidence of ET ranges from 0.59 to 2.53/100 000 inhabitants and its prevalence is around 30/100 000, which is similar to that of polycythemia vera (PV).25 The median age at diagnosis is 65–70 years,26 but ET is occasionally found in children, and it is relatively common in female patients in their third or fourth decade of life.27,28 In women of childbearing age, ET may be a risk factor for complications during pregnancy.29 Venous thrombosis in unusual locations is typical in younger patients, and the thrombotic risk increases in patients over 60 years old.26
Clinical features
Many patients are asymptomatic when an excess in platelets is discovered by a routine blood count.26,28,29 Most investigators have argued that the use of a threshold level 600 × 109/l platelets compromises the detection of early-phase disease because the adjusted 95th percentile for normal platelet count is below 400 × 109/l.30,31 Therefore, the platelet threshold count required for ET diagnosis has been lowered to 450 × 109/l (Box 23.1). Initial presentation may include vascular occlusions, hemorrhage or microvascular complications that lead to transient ischemic attacks and digital ischemia with paresthesias.32,33 Some patients may present with a thrombosis of major arteries and veins such as splenic or hepatic vein thrombosis as in the Budd–Chiari syndrome.26,34,35 Increase in spleen size at time of diagnosis is seen only in a minority of patients.2 Generally, there is no anemia, no circulating blasts, and serum levels of lactate dehydrogenase (LDH) are within the normal range.23,36 In patients with borderline anemia, palpable spleen, slight elevation of LDH serum level or evidence of erythroid and/or granulopoietic precursors in the PB, a differential diagnosis of very early (prefibrotic) stages of PMF should be excluded by careful bone marrow trephine biopsy (BMTB) examination.1,2,21,23,24,36,37 In contrast to PV cases, the utility of JAK2V617F mutation screening for the diagnosis of ET is limited by suboptimal negative predictive value and lack of diagnostic specificity in the context of MPN.14 Therefore, a BMTB is mandatory to differentiate ET from other MPN and to help with the differential diagnosis between JAK2V617F-negative ET and reactive thrombocytosis.1,14,21,24
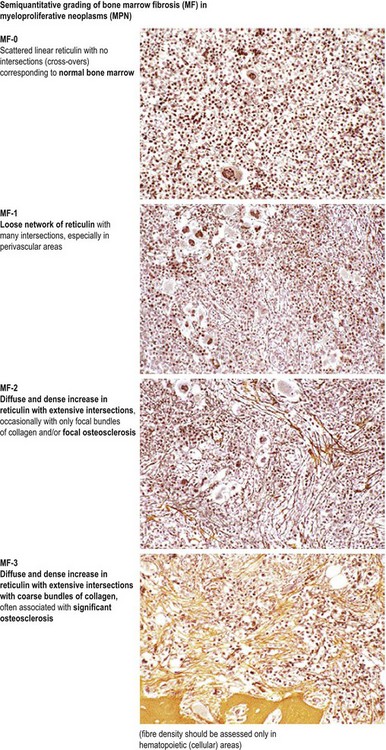
In most patients, ET is an indolent disorder characterized by long symptom-free intervals, interrupted by occasional life-threatening thromboembolic or hemorrhagic episodes.26,28,38 Although after many years a few patients with ET may develop BM fibrosis such progression is very uncommon.39–41 The exact incidence of myelofibrotic transformation in ET persists to be a controversial issue42 and the reported results are certainly influenced by the risk status of the patients and the applied diagnostic criteria.1,21,43,44 Careful morphological examination of BMTB is crucial in the diagnostic workup of post ET myelofibrosis (Post-ET MF), since appearance of BM fibrosis with a grade MF-2 or MF-3 is a prerequisite for the diagnosis (Box 23.2 and Fig. 23.1). Secondly, only cases with predefined and documented ET according to the WHO guidelines fall into this category.45 Strict adherence to the WHO criteria (Box 23.1) is necessary to prevent diagnostic confusion associated with early PMF accompanied by thrombocytosis.1,23,36 The frequency of true post-ET myelofibrosis is low; the transformation rate is 2.8% at a median follow-up of 9.1 years (or a 10-year risk of 3.9% and a 15-year risk of 6%).46 Transformation of ET to acute myeloid leukemia or MDS occurs in fewer than 5% of patients, and in chemotherapy treated patients may be related to previous cytotoxic therapy.26,28,47–49 The life expectancy in ET is near normal for most patients.2,50,51
Box 23.2 WHO criteria for the diagnosis of post-essential thrombocythemia myelofibrosis (Post-ET MF)
Molecular data
A JAK2V617F or a functionally similar mutation may be detected in approximately 40–50% of patients (see Chapter 21 for details).5,52–55 However, these findings are not specific for ET and are also seen in other MPN subtypes. It has to be emphasized that significant differences in the JAK2V617F allele burden were described between ET and early PMF.4,6,56 As an independent marker this feature validates very nicely corresponding BMTB findings and thus supports the concept of differentiating ET from early PMF.1,21,23,24,57 A gain-of-function mutation of MPL has been reported in approximately 1–3% of patients with ET.58,59 None of these mutations are found in cases of reactive thrombocytosis. Although ET is usually characterized by a normal karyotype, an isolated del(5q) has been reported in few cases with ET. However, it is more likely that these cases represent MDS associated with this abnormality.60,61
Blood and bone marrow findings
Marked and sustained thrombocytosis is the hallmark of ET. The platelets often display anisocytosis, ranging from tiny forms to atypical large, giant platelets that may reveal bizarre shapes, pseudopods and agranularity. The white blood cell count (WBC) and leukocyte differential are usually normal, although a borderline elevation in the neutrophil lineage may occur.28,62,63 In ET classified according to the histological guidelines of the WHO classification, neither a relevant increase in cellularity nor a significant left-shifted neutrophil granulopoiesis is observed.1,3,57 Any case with a mild to moderate granulocytic and erythroid growth pattern (panmyelosis) and an EPO level below the reference range is suspicious for occult (pre-polycythemic) PV mimicking ET.64,65 Regarding megakaryopoiesis, gross disturbances of the histological topography (significant abnormal localization and/or extensive dense clustering) are not seen (Figs 23.2 and 23.3). Megakaryocytes show a more or less random distribution, with scattered forms or a few loose clusters (Figs 23.4 and 23.5). A predominance of large to giant mature megakaryocytes with extensively folded (staghorn-like) nuclei19,20,22,24,66,67 surrounded by a correspondingly mature cytoplasm is found (Fig. 23.6). These features are clearly different from prefibrotic early PMF, where megakaryocytes show an extensive dense clustering and have hypolobulated (cloud-like or bulbous) and hyperchromatic nuclei with striking maturation defects (Fig. 23.7) leading to a marked anomaly of their nuclear-cytoplasmic ratio.1,19–22,24,36,65,67 Finally, there is no substantial increase in reticulin fibers at presentation and collagen fibrosis is never observed in ET.44 In a large series of ET patients, minimal to slight reticulin fibrosis was described in only 3% of cases.19,40,41,68,69 Any marked increase in reticulin fibers is not compatible with ET,1 in contrast to prefibrotic PMF, which is a differential diagnosis in patients with thrombocytosis.2,21,22,57 In PMF cases, a relevant increase in age-matched cellularity and a significantly expressed left-shifted neutrophil granulopoiesis is regularly found. At times, the clinical and morphological distinction between ET and early phase PMF with associated thrombocytosis might not be clear cut. As the therapeutic relevance in these early stages is still unclear, a strict adherence to the WHO criteria for making a working diagnosis and close monitoring of the patient to capture any substantial changes that might warrant revision of diagnosis is recommended.3,11,15
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


