CHAPTER 8 Erythroenzyme disorders
Introduction
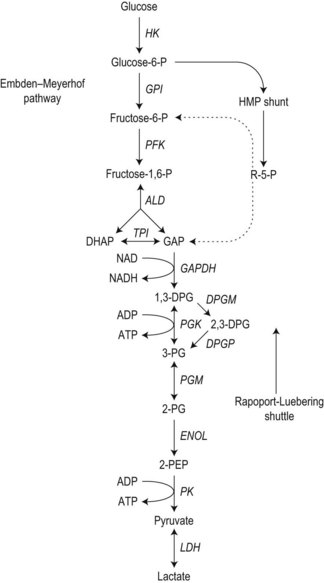
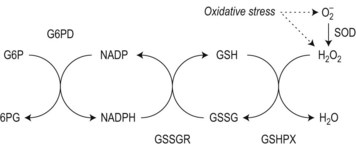
To achieve optimal performance as an oxygen transporter the mature red cell has sacrificed metabolic versatility.1 Red cell structural and functional integrity depend on catabolism of glucose via the anerobic Embden–Meyerhof pathway to replenish adenosine triphosphate (ATP) required for cation homeostasis and other energy-dependent processes in conjunction with the oxidative pentose phosphate pathway (hexose monophosphate shunt) to maintain redox capacity. In the resting state 90% of glucose is catabolized anerobically through the Embden–Meyerhof pathway, which also serves to generate nicotinamide adenine dinucleotide in its reduced form (NADH) required as a cofactor for cytochrome b5 reductase for the conversion of methemoglobin to hemoglobin (Fig. 8.1). The pentose phosphate pathway serves mainly to supply the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) necessary to regenerate reduced glutathione (GSH), which acts as a sacrificial reductant to protect the membrane and contents of the red cell against oxidative damage (Fig. 8.2). Glucose-6-phosphate dehydrogenase (G6PD) catalyzes the first and rate limiting step, the conversion of glucose-6-phosphate (G6P) to 6-phosphogluconate (6PG), in this pathway. Synthesis of the tripeptide glutathione from its constituent amino acids and nucleotide salvage complete the essential metabolic repertoire active in the red cell cytosol. Conservation of adenine nucleotides to maintain intracellular ATP and elimination of pyrimidine nucleotides is facilitated through the action of pyrimidine 5′-nucleotidase (P5N) which specifically dephosphorylates pyrimidine nucleoside-5′-monophosphates formed by RNA breakdown. This permits removal of toxic pyrimidines by passive diffusion and prevents their accumulation within the red cell. Defects in each of these key pathways produces hemolytic anemia.
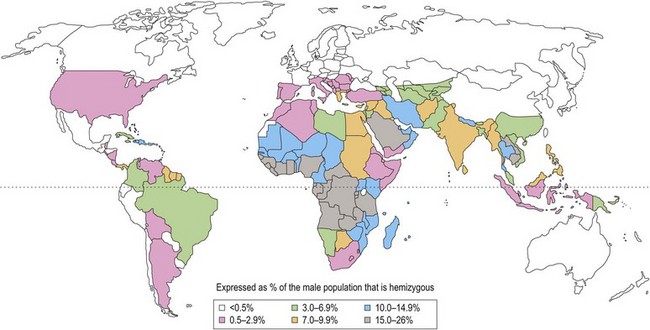
With the exception of polymorphic G6PD variants (Fig. 8.3) estimated to affect up to 400 million people worldwide, most inherited disorders of red cell metabolism are uncommon. Pyruvate kinase (PK) deficiency is the most commonly encountered defect of the Embden–Meyerhof pathway with around 500 cases reported to date. Heterozygote frequencies based on biochemical population studies vary from 0.14% in the US2 to 6% in Saudi Arabia.3 There is some evidence that reduced red cell PK activity affords protection against malaria.4 Mice deficient in PK are protected from malaria and the growth of Plasmodium falciparum is impaired in PK-deficient red cells. The impact of this on populations in areas of malarial endemicity is, however, likely to be small compared with the selective advantage conferred by the more common genetic red cell variants, G6PD deficiency and the hemoglobinopathies.
After PK the most common enzyme deficiencies implicated in hemolytic anemia are in approximate order of frequency: glucosephosphate isomerase (GPI); class I G6PD variants (associated with chronic hemolytic anemia); phosphofructokinase (PFK), triosephosphate isomerase (TPI), phosphoglycerate kinase (PGK) and hexokinase (HK). Deficiencies of P5N,5 which has been described in populations of diverse geographic origin, and glutathione synthetase6 occur at a comparable frequency. Other erythroenzyme disorders are rare.
Clinical features
The presence of neurological, myopathic or other non-hematologic manifestations is of considerable diagnostic value. Neurodevelopmental abnormalities must be interpreted with caution since severe neonatal hyperbilirubinemia sufficient to cause kernicterus has been described in deficiency of several red cell enzymes including G6PD and PK. Distinctive features of erythroenzymopathies are summarized in Table 8.1.
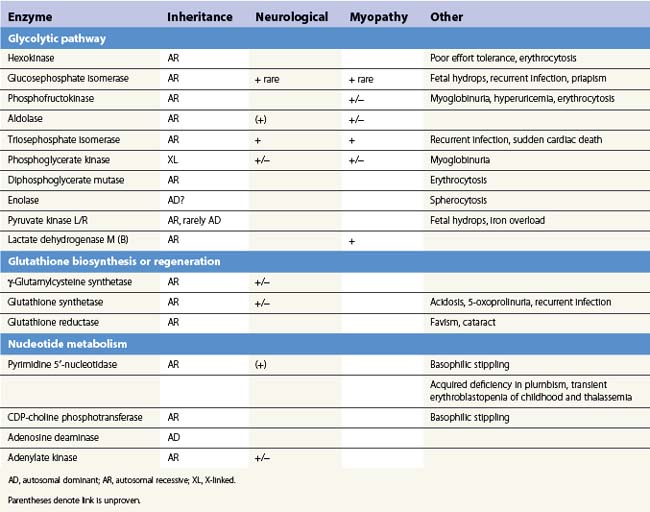
The pattern of somatic abnormalities manifest in individual enzyme disorders is determined by several factors. Deficiency of an isoenzyme, expression of which is restricted (e.g. pyruvate kinase-L/R), generally causes isolated hemolysis. Conversely, defects of ubiquitously expressed enzymes (e.g. triosephosphate isomerase) often result in a more generalized phenotype. The physiochemical properties of a mutant enzyme also influence clinical expression. Enzyme variants associated with impaired catalytic efficiency generally produce greater metabolic perturbance than those resulting in structural instability, the consequences of which are offset in tissues which retain the capacity for protein synthesis. This is reflected in a correspondingly more severe clinical phenotype. Examples include stable GPI mutants associated with multisystem7 disease and high Km class I G6PD variants which in addition to hemolysis manifest impaired leukocyte function or cataract due to reduction of enzyme activity in non-erythroid tissues.8
Blood cell morphology
Red cell enzyme defects have traditionally been grouped under the heading congenital non-spherocytic hemolytic anemia. While morphological atypia in the red cells are usually apparent, these overlap with other causes of hemolysis and are seldom exclusive to a specific enzyme defect. A notable exception is the striking basophilic stippling associated with P5N deficiency which may be seen in up to 5% of red cells on a freshly stained blood film prepared from an ethylenediaminetetraacetic acid (EDTA) sample (Fig. 8.4). Stippling may disappear if the stain is delayed by more than a few hours, presumably because EDTA chelates metal ions required for ribonucleoprotein aggregation. This problem may be circumvented by examination of blood taken into lithium heparin. Conspicuous punctate basophilia akin to that in P5N deficiency accompanies unstable hemoglobin variants and CDP-choline phosphotransferase deficiency,9 a putative defect of nucleotide metabolism described in only a few families. Lead is a potent inhibitor of P5N activity. This explains the finding of punctate basophilia in plumbism. The presence of other clinical and morphologic stigmata, the latter including red cell hypochromia and microcytosis, reticulocytosis or sideroblastic erythropoiesis, usually render distinction from primary P5N deficiency straightforward. Basophilic stippling may also be found in a wide range of congenital and acquired dyserythropoietic states including thalassemia and occasionally in some glycolytic defects (e.g. pyruvate kinase or phosphofructokinase deficiency).
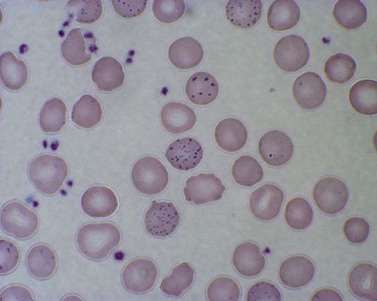
Fig. 8.4 Coarse basophilic stippling in pyrimidine 5′-nucleotidase deficiency. May–Grünwald–Giemsa. × 1000.
Features of oxidative damage to red cells are most commonly associated with, though not confined to, G6PD deficiency. These are most remarkable during hemolytic crises following exposure to oxidant drugs (Table 8.2) or fava bean (broad bean) consumption and include irregularly contracted hyperchromic erythrocytes, some of which display a characteristic ‘bite’ or ‘hemighost’ appearance (Fig. 8.5). ‘Bite’ cells in which the surface of the erythrocyte is breached producing an irregular gap are thought to be generated by removal of Heinz bodies during transit through the spleen. Erythrocyte ‘hemighosts’ are forms in which the hemoglobin appears condensed and is retracted to one side leaving an empty space in the cell. In the common polymorphic G6PD variants (e.g. G6PD A− or Med) these morphologic abnormalities are visible only during hemolytic episodes. Following acute hemolysis in G6PD deficiency rapid clearance of damaged cells by the spleen ensues and during the recovery phase polychromasia and macrocytosis predominate. Similar though usually less marked features of oxidative damage may be seen in defects which impair glutathione biosynthesis (Fig. 8.6) or regeneration due to deficiency of γ-glutamylcysteine synthetase,10 glutathione synthetase6,11 or glutathione reductase12 as well as neonatal hemolysis due to deficiency of glutathione peroxidase (Fig. 8.7).13 The latter condition which does not conform to the laws of Mendelian inheritance may reflect a transient reduction in enzyme activity due to impaired selenium homeostasis in the mother or neonate. Selenium is an essential co-factor for glutathione peroxidase which serves to detoxify harmful peroxides in the red cell. Glutathione peroxidase deficiency produces an acute and usually self-limiting hemolytic anemia in the newborn period. The diagnosis may be confirmed by assay of neonatal and maternal selenium levels and red cell glutathione peroxidase. Defects of other enzymes in the pentose phosphate pathway, 6-phosphogluconolactonase14 and phosphogluconate dehydrogenase15 have been described, albeit rarely, and should be considered if changes suggestive of oxidative damage are evident and more common causes excluded.
Table 8.2 Drugs and chemicals associated with hemolysis in glucose-6-phosphate dehydrogenase (G6PD) deficiency
| Class of drug | Examples |
|---|---|
| Antimalarials | Primaquine, pentaquine, pamaquine, chloroquine* |
| Sulfonamides and sulfones | Sulfanilamide, sulfacetamide, sulfapyridine, sulfamethoxazole (including co-trimoxazole), dapsone |
| Other antibacterial agents | Nitrofurantoin, nalidixic acid, chloramphenicol, ciprofloxacin* |
| Analgesic/antipyretic | Acetanilid, acetylsalicylic acid (aspirin)†, paracetamol (acetaminophen)† |
| Miscellaneous | Probenecid |
| Dimercaprol | |
| Vitamin K analogs | |
| Naphthalene (moth balls) | |
| Methylene blue | |
| Ascorbic acid | |
| Trinitrotoluene |
† Only after high doses or overdose.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


