CHAPTER 22 Erythrocytosis and polycythemia
Introduction
The term ‘erythrocytosis’ is derived from Greek words meaning ‘too many red cells’ and should be distinguished from ‘polycythemia’, meaning ‘too many cells in the blood’.1 Erythrocytosis has been defined as a greater than two standard deviation-increase from the age-, sex- and race-adjusted norm in hematocrit or hemoglobin level.2 It is clear, however, that these laboratory parameters may be affected by decreases in plasma volume. Therefore, a clinical diagnosis of ‘erythrocytosis’ might represent true erythrocytosis, indicating a true increase in red cell mass (RCM), or in fact apparent polycythemia, resulting from either reduced plasma volume (relative polycythemia) or failure to recognize otherwise normal values for hematocrit (Hct) or hemoglobin (Hgb) level that lie in the extreme right tail of the Gaussian distribution.3 Hence, for an individual patient, interpretation of laboratory results without knowledge of the personal baseline value remains inaccurate because of the inevitable statistical overlap of extreme values between subjects with and without disease.3
The recent development of molecular tests for the JAK2V617F and MPL mutations (see Chapter 21 for details) has allowed the reliable distinction of clonal from non-clonal erythrocytosis and has radically altered the investigation of erythrocytosis. Furthermore the value of aggressive phlebotomy to lower Hct levels below 45% in men and 42% in women has not been substantiated.3 The therapeutic relevance of distinguishing polycythemia vera (PV) from so-called ‘essential thrombocythemia (ET) with borderline increased Hct’ has diminished and has further undermined the value of RCM measurement, which is no longer fundamental to the diagnosis of many patients.3
In this chapter, the recent revision of the WHO criteria,4 which reflects developments in molecular pathogenesis and international consensus on clinico-pathological classification, is used for definition purposes although it is recognized that dissenting views have been published.5 Erythrocytosis is therefore practically defined according to the thresholds used in the WHO classification,3,4 i.e.:
The classification of erythrocytosis has recently been the subject of two excellent reviews.3,6 The term ‘idiopathic erythrocytosis’ was used by McMullin6 but has been criticized by Patnaik and Tefferi3 as an entity, due to misuse of the term for patients who have an inappropriate diagnosis of erythrocytosis or who have been inadequately investigated. Nevertheless, there remain patients who have been fully investigated and who are likely to have abnormalities that have not as yet been defined. These may include defects of the erythropoietin (Epo) signaling pathway or oxygen sensing pathway.3,6 Therefore, the category ‘unclassifiable’ is introduced here in accordance with the WHO classification and strictly defined for patients who have been fully investigated and for whom no currently defined cause of the erythrocytosis has been found. Patients who are partially investigated should not be placed in this category. While there are several potential divisions of a classification of patients with proven erythrocytosis, the classification used here (Box 22.1) is adapted from Patnaik and Tefferi.3
The pathology of erythrocytosis
The discovery of the JAK2V617F mutation in 20057–10 (see Chapter 21) has profoundly changed both understanding and investigation of the erythrocytoses. Accordingly, modern diagnostic evaluation of a patient with proven erythrocytosis, and for whom no obvious cause is apparent, may begin with screening of the peripheral blood for the JAK2V617F mutation and serum Epo. A working classification of erythrocytosis into primary, or clonal, i.e. PV, versus secondary has therefore emerged,6 but the more classical pathogenetic approach is used here.
Congenital erythrocytosis
Associated with reduced P50
High-oxygen-affinity hemoglobinopathy
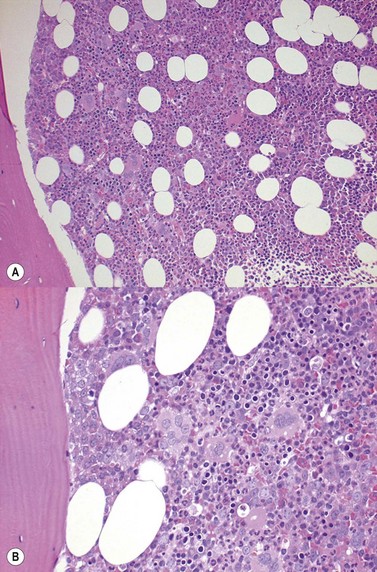
High-oxygen-affinity hemoglobins release oxygen at a lower rate than normal and thus create relative tissue hypoxia, which might result in compensatory erythrocytosis in approximately one third of affected patients. Affected patients often present with isolated erythrocytosis, in the absence of signs and symptoms of systemic disease.3 Erythrocytosis is accompanied by chronic hemolysis where the Hgb variant is unstable.6 Bone marrow trephine biopsy (BMTB) investigation shows erythroid hyperplasia but normal megakaryocytes (Fig. 22.1A, B).These patients may have family members who are similarly affected and a family history is therefore essential in the investigation of patients with erythrocytosis. Transmission in affected patients is usually autosomal dominant.
More than 90 mutations have been described and are covered in an exemplary fashion in a web resource: http://globin.bx.psu.edu/hbvar/. Most of the high-oxygen-affinity mutations involve the β-globin chain and α1β2 contact zones.11 Serum Epo levels are either normal or elevated and P50 (partial pressure of oxygen at which 50% of Hgb is saturated with oxygen) is decreased.12 Structurally abnormal high-affinity Hgb should be suspected3 if the P50 level is <20 mmHg.
2,3-Bisphosphoglycerate mutase (BPGM) deficiency
BPGM deficiency is a rare cause of erythrocytosis.13,14 Deficiency of the enzyme results in a high affinity Hgb with a left shifted oxygen dissociation curve. This results in a compensatory erythrocytosis. Patients with both autosomal dominant15 and autosomal recessive inheritance16 have been described. In a fully penetrant autosomal recessive case, there is an isolated erythrocytosis with normal serum Epo level.16 Diagnosis is established by showing a low P50, a normal Hgb structure and decreased BPGM activity.3
Methemoglobinemia
Congenital methemoglobinemias are of three main types:17,18
Methemoglobin causes both an impaired O2 binding and increased oxygen affinity of the Hgb; sometimes resulting in compensatory erythrocytosis.18
Methemoglobinemia is clinically suspected when cyanosis is accompanied by normal PaO2 levels but low saturation per pulse oximeter. Both carboxyhemoglobin and methemoglobin may be measured by modern oximeters.3
Associated with normal P50
The involvement of oxygen-sensing pathways in physiologic and pathologic erythropoiesis has recently been reviewed.20 Red blood cells deliver O2 from the lungs to the tissues. Better understanding of the physiological regulation of the process has allowed a rational classification of rare cases of congenital erythrocytosis to be developed (Fig. 22.2).3,6
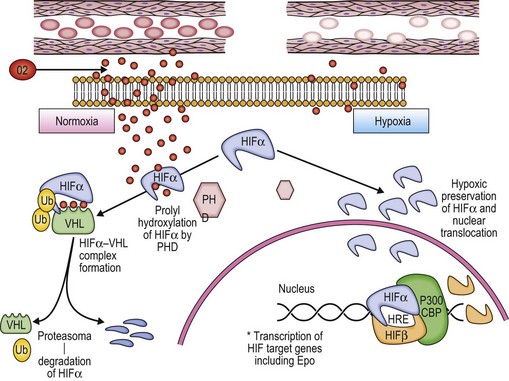
A classic physiologic response to hypoxia in humans is the up-regulation of the Erythropoietin (Epo) gene, which is the central regulator of red blood cell mass. Reduction of tissue oxygenation triggers increased production of Epo by hypoxia-inducible factor 1 (HIF-1), which is a transcriptional activator composed of an O2-regulated α subunit and a constitutively expressed β subunit. Hydroxylation of HIF-1α or HIF-2α by the asparaginyl hydroxylase FIH-1 blocks coactivator binding and transactivation. Hydroxylation of HIF-1α or HIF-2α by the prolyl hydroxylase PHD2 is required for binding of the von Hippel–Lindau (VHL) protein, leading to ubiquitination and proteasomal degradation. Mutations in the genes encoding VHL, PHD2, and HIF-2α have all been identified in patients with familial erythrocytosis.20
VHL mutations including Chuvash erythrocytosis (frequently termed Chuvash polycythemia)
Chuvash erythrocytosis is a rare, autosomal recessive, congenital erythrocytosis, first described in the Chuvash autonomous region in Russia21 but also occurring in other racial and ethnic groups.22,23 Affected patients are homozygous for a germline mutation affecting the VHL tumor suppressor gene, producing an abnormal VHL protein.24,25 The mutation in the VHL gene disrupts the normal mechanism of hypoxia sensing, ultimately resulting in increased Epo production and erythrocytosis.6
In contrast to patients with the VHL disease, an autosomal dominant familial syndrome, patients with Chuvash erythrocytosis do not display an increased predilection for tumors.3 However, other abnormalities are found.26 Plasma concentrations of endothelin-1, Epo, plasminogen activator inhibitor-1, transferrin, transferrin receptor, and vascular endothelial growth factor are elevated. Clinical manifestations include increased cardiac valvular abnormalities, hemangiomas, pulmonary arterial hypertension, thrombotic and hemorrhagic events, varicose veins, and shortened life span. In parallel, peripheral blood concentrations of CD4 positive T-helper cells and CD4/CD8 ratio have been found to be lower in the VHL598C>T homozygotes.26
Prolyl hydroxlase domain 2 (PHD2) mutations
Under normoxic conditions, PHD2 hydroxylates the α-subunits of HIF proteins, facilitating VHL binding and subsequent ubiquitin-mediated proteasomal degradation of HIF.3,6 Rare cases of PHD2 mutations have been described, with the loss of PHD2 function, associated erythrocytosis and normal serum Epo.3,6 Recently, a novel PHD2 mutation has been described, associated with both erythrocytosis and recurrent paraganglioma.27
Hypoxia inducible factor alpha (HIF2α) mutations
Several erythrocytosis-associated HIF2α mutations have been characterized. All result in impaired degradation and thus aberrant stabilization of HIF2α. However, each exhibits a distinct profile with respect to their effects on PHD2 binding and VHL interaction.28 Epo levels are usually elevated.3,6
Epo receptor (Epo-R) mutations
Epo-R signaling is regulated by the binding of the protein tyrosine phosphatase SHP-1 (or other JAK/STAT regulators) to the distal cytoplasmic region of Epo-R. This interaction results in the down-regulation of the Epo-mediated activation of the JAK2/STAT5 pathway and ultimately production of more red cells.3,6 Epo-R mutations (all in exon 8) that cause erythrocytosis have been reviewed.29–31 These mutations often result in cytoplasmic truncation of Epo-R, resulting in failure of attachment of SHP-1, ongoing production of red cells and thus erythrocytosis.
Transmission is usually autosomal dominant. Affected patients are usually asymptomatic and display subnormal (or normal) serum Epo level and hypersensitivity of erythroid progenitors to exogenous Epo.3,6
Acquired erythrocytosis
Erythrocytosis secondary to hypoxia
Chronic hypoxia leads to physiological secondary erythrocytosis, in order to compensate for the low oxygen concentrations at the pulmonary and/or tissue level. Clinically, interpretation of arterial blood gases, pulmonary function tests and chest radiography is an integral part of the investigation of erythrocytosis.3 Chronic lung disease, right-to-left cardiopulmonary shunts, high-altitude habitat, tobacco use/carbon monoxide poisoning, sleep apnea/hypoventilation syndrome and renal artery stenosis are all associated with secondary erythrocytosis.3,6 The bone marrow (BM) morphology is similar to that of patients with congenital erythrocytosis, showing hyperplasia of erythropoiesis and normal megakaryocytes (Fig. 22.1).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


