Enteropathogenic Escherichia coli
Shahista Nisa, Karen M. Scanlon and Michael S. Donnenberg, University of Maryland School of Medicine, Baltimore, MD, USA
Background
Definition and classification
Enteropathogenic E. coli (EPEC) strains are diarrhea-causing, non-Shiga-toxin-producing E. coli. In healthy adults, EPEC-induced diarrhea may be initiated by a dose of 108 to 1010 CFU, though this infectious dose is likely lower for children (Donnenberg et al., 1998). Upon intimate attachment, these strains efface the host cell microvilli, generating a characteristic histopathology known as the attaching and effacing (A/E) effect. In these ways, EPEC is distinguished from other diarrheagenic pathotypes, as enteroaggregative E. coli (EAEC), enteroinvasive E. coli (EIEC), and enterotoxigenic E. coli (ETEC) do not display an A/E effect, while enterohemorrhagic E. coli (EHEC) express Shiga toxins (Nataro and Kaper, 1998). The genetic elements responsible for the production of A/E pathology are harbored on a large pathogenicity island known as the locus of enterocyte effacement (LEE) (McDaniel and Kaper, 1997). In addition, EPEC strains are further classified as typical or atypical by the presence or absence of an EPEC adherence factor (EAF) plasmid encoding the BFP.
History
In the 1930s, serological methods were used to characterize E. coli infections among both infants and calves, but the significance of E. coli in diarrheal disease was not fully appreciated until 1945 (Taylor, 1961). Bray associated specific E. coli, later recognized as serogroup O111, with an outbreak of infantile diarrhea, and this was quickly succeeded by a number of publications which further established the importance of O111 and extended the implicated sero-groups to include O55, O26, and O119 (Bray, 1945). In the 1950s, with the advent of an internationally accepted serological testing method, it was soon reported that EPEC had a worldwide distribution. For decades thereafter, EPEC was described solely based on the O and H serotypes prevalent amongst children under 2 years with diarrhea (Robins-Browne, 1987). WHO recognizes 12 EPEC serotypes, including typical and atypical EPEC strains, along with other diarrheagenic E. coli, such as EAEC and EHEC (Campos et al., 1994; Rodrigues et al., 1996; Scotland et al., 1996; Do Valle et al., 1997; Trabulsi et al., 2002). However, it is now appreciated that assessments of genotype and phenotype, and not serotype, are more accurate methods for identifying EPEC. The use of these descriptors has shed new light on the epidemiological and evolutionary features of this pathogen, in addition to bringing about a change in the classification of certain strains.
Epidemiology and global impact
EPEC is a major cause of human diarrhea in the developing world; with rare outbreaks in developed countries typically associated with nurseries and day-care centers (Donnenberg and Kaper, 1992; Nataro and Kaper, 1998). Adult infections are uncommon and typically the result of common source outbreaks or concordant with other conditions (Nataro and Kaper, 1998). Previously, rates of community-acquired EPEC infection were thought to be highest within the first 6 months after birth (Chatkaeomorakot et al., 1987; Cravioto et al., 1988; Gomes et al., 1991), but recent studies of children 5 years and younger indicate that prevalence increases with age (Behiry et al., 2011; Ochoa and Contreras, 2011). The disparity observed among publications describing age-associated incidence of infection may be due to changes in EPEC diagnosis, promotion of breastfeeding, or evolution of the pathogen. Disease severity, however, remains inversely proportionate to age (Toledo et al., 1983; Cravioto et al., 1990). In addition to age, it has been proposed that bacterial load may be a factor in EPEC-induced diarrhea (Barletta et al., 2011). The described incidence of infection differs greatly among studies, with EPEC-associated diarrhea ranging from 3–50% (Gomes et al., 1991; Alikhani et al., 2006; Nweze, 2010). Emergent reports, in which EPEC was characterized genetically and not by serotype, indicate a high prevalence of atypical EPEC (aEPEC), particularly in developed regions (Afset et al., 2003; Robins-Browne et al., 2004; Liebchen et al., 2011) and associated prolonged diarrhea with these strains (Afset et al., 2004; Nguyen et al., 2006). While humans are the sole reservoir for typical EPEC (tEPEC), animals may act as reservoirs for aEPEC; this chapter will focus on those typical and atypical strains affecting humans.
Molecular pathogenesis
Regulation
Over the years, multiple proven and potential virulence factors that help facilitate EPEC colonization of the intestinal epithelium have been identified. The expression, secretion, and translocation of effector proteins that enable the bacteria to subvert host cellular pathways and cause disease are tightly regulated. These virulence factors are regulated by elements encoded on the EAF plasmid as well as the LEE pathogenicity island.
The EAF plasmid
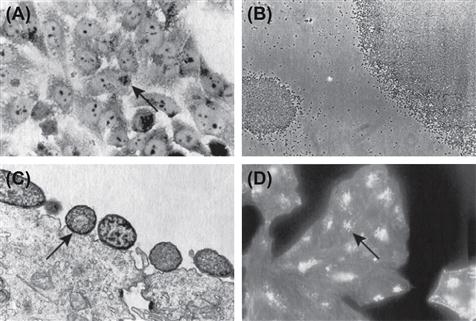
The large (50–70 MDa) EAF plasmid harbored by tEPEC strains is required for localized adherence (LA; Figure 4.1A) (Baldini et al., 1983; Nataro et al., 1985; McConnell et al., 1989). Curing the EAF plasmid from EPEC abolished LA and autoaggregation phenotypes (Figure 4.1B) (Baldini et al., 1983; Knutton et al., 1987a; Chart et al., 1988) and led to attentuated virulence in colostrum-deprived piglets (Baldini et al., 1983) and healthy volunteers (Levine et al., 1985). However, plasmid-cured EPEC retained the ability to form A/E lesions (Figure 4.1C) (Baldini et al., 1983; Knutton et al., 1987a,b; Tzipori et al., 1989) and transfer of the EAF plasmid to non-EPEC strains endowed them with the ability to carry out LA (Baldini et al., 1983; Knutton et al., 1987b).
Sequence analysis of the EAF plasmid from E2348/69, pMAR7, a modified pMAR2 plasmid that carries an ampicillin resistance marker, revealed it to be a self-transmissible plasmid due to the presence of the highly conserved tra locus (Brinkley et al., 2006). The complete tra region is only found in representatives of the EPEC1 clonal group (Tobe et al., 1999; Brinkley et al., 2006), confirming previous findings that some EAF plasmids can be mobilized to other E. coli strains by conjugation (Baldini et al., 1983; Laporta et al., 1986; McConnell et al., 1989) while others cannot be transmitted as they lack the required transfer genes (Hales et al., 1992; Tobe et al., 1999).
Two loci important for pathogenicity on the EAF plasmid are the type IV BFP gene cluster (bfp) and the per locus encoding for transcriptional activators. The EAF plasmid also regulates the expression of the eae gene, located in the chromosomal pathogenicity island LEE (Jerse and Kaper, 1991) (see below).
Bundle-forming pili
BFP is a member of the type IV-B class of fimbriae that are produced by a number of Gram-negative pathogenic bacteria (e.g. ETEC, Salmonella enerica serovar Typhi and Vibrio cholerae). BFPs form ropelike bundles extending from the cell surface of typical EPEC strains (Girón et al., 1991). These pili are important virulence factors and are required for LA to epithelial cells (Donnenberg et al., 1992a), autoaggregation and virulence in volunteers (Bieber et al., 1998; Knutton et al., 1999).
The bfp operon consists of 14 genes, all of which are required for BFP biogenesis (Ramer et al., 1996; Sohel et al., 1996; Stone et al., 1996; Anantha et al., 2000; Blank and Donnenberg, 2001; Schreiber et al., 2002; Hwang et al., 2003). Expression of this gene cluster in a laboratory strain of E. coli via a plasmid confers both LA and BFP biogenesis (Stone et al., 1996). BFP is composed of repeating subunits of bundlin, pilin monomers encoded by bfpA, the first gene in the bfp operon. bfpA encodes pre-bundlin, which is processed by the pre-pilin peptidase, encoded by bfpP, producing mature bundlin (Donnenberg et al., 1992a,b; Zhang et al., 1994). Mutation of the bfpA gene leads to loss of LA and autoaggregation (Donnenberg et al., 1992a; Bieber et al., 1998; Anantha et al., 2000).
Structural analysis of bundlin revealed that while EPEC and Vibrio cholerae show distinct differences in their monomer subunits and are unable to complement one another, they assemble into filaments with similar helical organization (Ramboarina et al., 2005). The genes in the bfp operon are regulated by environmental and host factors together with a plasmid encoded regulator, Per (Puente et al., 1996; Rosenshine et al., 1996a; Martínez-Laguna et al., 1999). For more detailed information on BFP and other type IV pili, see Chapter 13.
per (plasmid-encoded regulator)
The per operon consists of three ORFs, designated perA, perB, and perC, which are also known as bfpT, bfpV, and bfpW, respectively. perA, the first gene, codes for a protein that belongs to the AraC family of transcriptional activators. The C-terminal region of PerA contains a DNA-binding helix-turn-helix motif which is highly conserved between strains (Okeke et al., 2001). PerA activates the expression of bfpA as perA inactivation leads to the loss of bfpA expression and thus LA (Tobe et al., 1996; Okeke et al., 2001; Ibarra et al., 2003). Volunteer studies demonstrated the contribution of perA and bfpA to diarrhea, with reduced virulence displayed by mutant strains (Bieber et al., 1998). PerA also auto-activates its own promoter, binding to AT-rich regions upstream of the gene (Martínez-Laguna et al., 1999). This region is significantly homologous to the PerA binding site upstream of bfpA (Bustamante et al., 1998). PerA is closely related to VirF and Rns, the activator of the virulence gene regulatory cascade in Shigella flexneri and the activator of virulence genes in ETEC, respectively (Dorman, 1992). However, while VirF and Rns could be functionally substituted (Porter et al., 1998; Munson et al., 2001), complementation of virF and rns mutant strains was not observed with PerA and likewise a perA mutant could not be complemented with either VirF or Rns (Porter et al., 2004).
Apart from activating the EAF plasmid encoded genes, the per regulon has been shown to activate the expression of chromosomally encoded genes present at the LEE, including the eae gene (Gómez-Duarte and Kaper, 1995). By ensuring adequate expression of PerC, PerA indirectly activates Ler, the LEE transcriptional activator (see below) (Mellies et al., 1999; Bustamante et al., 2011).
The LEE pathogenicity island and the type 3 secretion/translocation system
The LEE is a 35.6 kb pathogenicity island. Introduction of this region into a non-pathogenic E. coli strain confers the ability to induce the A/E effect (Figure 4.1C) (McDaniel and Kaper, 1997). The EPEC LEE contains 41 ORFs (Elliott et al., 1998); encoding for an adhesin, the type 3 secretion system (T3SS), effector proteins, type 3 specific chaperones and transcriptional regulators. The LEE contains five polycistronic operons, LEE1 to LEE5. The principal function of the LEE is to form the T3SS apparatus, enabling EPEC to transport proteins across three membranes: the cytoplasmic and outer membranes of EPEC and the cytoplasmic membrane of the host cell to which the EPEC are attached (Jarvis et al., 1995). T3SSs are composed of more than 20 proteins that form a complex apparatus spanning both bacterial membranes and culminating in a needle that extends from the outer membrane (see Chapter 14 for details). The EPEC T3SS also includes the translocator proteins EspA, EspB, and EspD. Mutations of espA, espB, or espD result in strains that are unable to translocate effector proteins into host cells (Kenny et al., 1997; Knutton et al., 1998; Taylor et al., 1998; Wolff et al., 1998; Kenny and Jepson, 2000) and therefore unable to cause an A/E effect and reorganization of the host cell cytoskeleton (Donnenberg et al., 1990a,b, 1993a,b,c; Foubister et al., 1994a,b; Kenny et al., 1996; Abe et al., 1997, 1998; Kenny and Finlay, 1997; Lai et al., 1997; Nougayrède et al., 1999). EspB is required for virulence in volunteers (Tacket et al., 2000), while in a rabbit model, EspA, EspB, and EspD have each been shown to be required for virulence (Abe et al., 1998). EspA is a component of a prominent filamentous structure that connects EPEC to host cells (Knutton et al., 1998). It forms a sheath around the needle complex which is encoded by escF (Sekiya et al., 2001; Wilson et al., 2001). EspB and EspD are both localized to the host cell membrane after attachment and together, these proteins form the translocation pore (Knutton et al., 1998; Wolff et al., 1998; Wachter et al., 1999; Ide et al., 2001; Luo and Donnenberg, 2011). In addition, EspB has been shown to be an effector protein secreted by the T3SS. EspB binding to α-catenin (Kodama et al., 2002) and myosin (Iizumi et al., 2007) within the host cell regulates actin cytoskeletal rearrangement and thus promotes morphological changes (Taylor et al., 1999).
Recently, other proteins have been identified as being crucial for the T3SS. EscA, a small protein encoded by Orf15, localizes to the periplasm, associates with the inner membrane, interacts with outer membrane secretin EscC and is required for the structural integrity of the T3SS complex (Sal-Man et al., 2012a). EscI has also been shown to be essential for T3S and may function as an inner rod protein and act as an early secreted effector (Sal-Man et al., 2012b). EscI interacts with EscU, which is an essential inner membrane ring of the T3SS that self-cleaves to form a secretin-competent state for accommodating effectors (Zarivach et al., 2008; Thomassin et al., 2011). The EscF needle protein associates with multiple proteins: EscD, an inner membrane structural protein, EscJ, an inner ring protein, and EscC. EscC, EscD, and EscJ are required for the formation of the T3SS apparatus (Ogino et al., 2006). EscI, EscU, EscF, and the chaperone CesT also interact with EscP (Orf16) (Monjaras et al., 2012). While EscP is not an essential protein of the T3SS, it appears to be required for regulating needle length and participating in substrate specificity (Monjaras et al., 2012). EscN is a type III ATPase (Andrade et al., 2007) that provides a potential energy source for the unfolding of T3S effectors (Song et al., 2004; Zarivach et al., 2007).
Delivery of effectors by the T3SS leads to the interaction between the products of two highly characterized genes in the LEE, eae and tir, encoding intimin and Tir, respectively. Intimin is a 94-kDa outer membrane adhesin (Jerse and Kaper, 1991) required for the intimate attachment of EPEC to epithelial cells at the site of A/E lesions (Figure 4.1C) (Jerse et al., 1990), while Tir is the translocated intimin receptor which binds intimin at the host cell surface (Kenny et al., 1997). While intimin is not strictly required for EPEC protein secretion or translocation (Rosenshine et al., 1992; Foubister et al., 1994b; Haigh et al., 1995; Kenny and Finlay, 1995, 1997; Wolff et al., 1998), the absence of intimin results in a significant decrease in EspB translocation (Wolff et al., 1998) and intiminis required for full virulence in human and rabbit infection (Donnenberg et al., 1992b; Marchès et al., 2000). There are 29 subtypes of intimin described to date. These subtypes are differentiated by a variable C-terminal region comprising the last 280 amino acids that is required for intimin–Tir binding and controlling specific host and tissue tropism (Fitzhenry et al., 2002; Ito et al., 2007; Mundy et al., 2007). Intimin β is the most prevalent subtype found amongst isolates of EPEC from humans and animals (Ramachandran et al., 2003; Nakazato et al., 2004; Blanco et al., 2005). Mutation of the eae gene leads to loss of A/E and host cell invasion (Francis et al., 1991), in addition to preventing the promotion of pseudopod formation (Rosenshine et al., 1996b) and EPEC attachment to human mucosal explants (Hicks et al., 1998). Unlike T3SS mutants, eae mutants retain the ability to redistribute host cell actin but have a blunted capacity to concentrate host cell cytoskeletal elements beneath the attachment site (Donnenberg et al., 1990a; Rosenshine et al., 1992). Mutant eae strains of EPEC can complement strains carrying mutations in espA, espB or other T3SS genes, allowing them to establish A/E (Rosenshine et al., 1992; Donnenberg et al., 1993b; Kenny et al., 1996). In addition, pre-infection with an eae mutant allows a laboratory E. coli strain carrying the cloned intimin gene (Rosenshine et al., 1996a) or beads coated with intimin (Liu et al., 1999) to bind to epithelial cells. Such trans complementation is possible because the eae mutant remains capable of translocating Tir into the host cell membrane. Apart from binding Tir, intimin may also bind to host intimin receptors (Hir) such as nucleolin via its variable C-terminus (Mundy et al., 2007).
Tir is a multifunctional protein which, after injection into the host cell via the T3SS, becomes integrated into the host cell membrane in a protease-sensitive conformation (Rosenshine et al., 1996a; Kenny et al., 1997). This protein exhibits a dimeric hairpin topology, such that an extracellular loop from each monomer binds an intimin molecule (De Grado et al., 1999; Hartland et al., 1999; Kenny, 1999) with the N-terminal and C-terminal domains projecting into the host cell cytoplasm (Figure 4.2B). Tir serves as the focus for the actin reorganization that occurs at the site of EPEC pedestals (Figure 4.1D). Some, but not all EPEC strains express Tir variants that have a tyrosine at position 474 (Y474). Upon translocation of this form of Tir into the host cell, it is phosphorylated at Y474 (Kenny et al., 1997) by host cell kinases (Nakazato et al., 2004; Swimm et al., 2004; Blanco et al., 2005). This event is essential for actin remodeling in such strains (Kenny, 1999), as mutation of Tir results in the loss of the A/E phenotype and reduced virulence in a rabbit model (Kenny et al., 1997; Marchès et al., 2000). As discussed below, there are other pathways leading to indistinguishable actin remodeling.
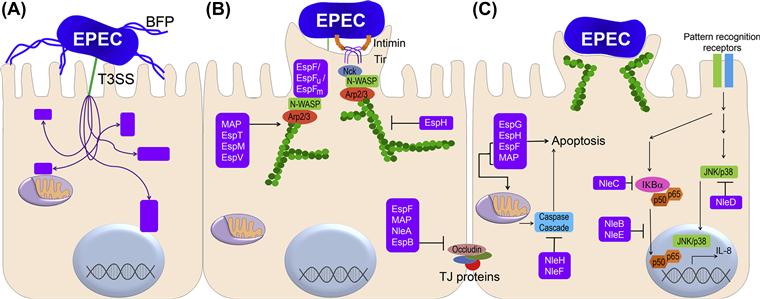
Apart from Tir, other translocated LEE encoded effector molecules include EspF, EspG, EspH, EspZ, and Map (Pallen et al., 2005). More recently, a repertoire of non-LEE encoded effectors (Nles) that are secreted by the T3SS have been discovered. For more detailed information on the type 3 effectors, see Chapter 15.
Ler (LEE-encoded regulator)
Ler is a 15-kDa protein encoded by the first gene in the LEE1 operon (Mellies et al., 1999). Ler regulates the expression of LEE as well as some non-LEE-encoded genes (Elliott et al., 2000). This protein induces expression by counteracting the repression mediated by nucleoid-associated protein H-NS (Bustamante et al., 2001). H-NS binds to AT-rich sequences and negatively modulates expression of a number of genes (Atlung and Ingmer, 1997). Expression of Ler is influenced by many factors in response to environmental signals (Mellies et al., 2007) including quorum sensing (Sperandio et al., 1999). Quorum sensing activates quorum-sensing E. coli regulator A (QseA), which activates transcription of Ler independent of per (Sircili et al., 2004). Ler expression is also dependent on integration host factor (IHF). IHF positively regulates ler by binding upstream of the ler promoter (Friedberg et al., 1999). GlrA (Global Regulator of LEE-Activator) and GrlR (Repressor) encoded within the LEE positively and negatively regulate Ler expression respectively (Deng et al., 2004). PerC, encoded in the EAF plasmid, has also been shown to induce the expression of Ler (Lio and Syu, 2004), independent of activation via GlrA (Bustamante et al., 2011). Ler is also a negative auto-regulator (Berdichevsky et al., 2005). It binds to the LEE1 regulatory region to reduce Ler, enabling the activation of the LEE2, LEE3, and other promoters.
Other virulence factors
Lymphostatin/Efa1
The lifA gene, encoding a 366-kDa protein lymphostatin, is located on the IE6 pathogenicity island. This pathogenicity island also encodes several non-LEE-encoded effectors (Iguchi et al., 2009). To date, lymphostatin is the largest T3S effector protein identified (Deng et al., 2012), both in EPEC and EHEC. The prevalence of this locus in EHEC strains isolated from patients with hemolytic uremia and outbreak strains suggests it may play a role in virulence (Afset et al., 2006; Wickham et al., 2006). A portion of lymphostatin displays sequence similarity to the glycosyltransferase region of large clostridial cytotoxins, which is required for glycosylation of the Rho family GTPases (Klapproth et al., 2000). A lifA mutant of Citrobacter rodentium was deficient in decreasing epithelial barrier function. This change was dependent on the glycosyltransferase region and was associated with a decreased activation of GTPase Cdc42 (Babbin et al., 2009). Multiple functions have been described for this extremely large protein. C. rodentium lymphostatin is also involved in bacterial colonization, crypt cell proliferation, and epithelial cell regeneration (Klapproth et al., 2005). In EPEC, lymphostatin inhibits the transcription of many lymphokines, including interleukin 2, interleukin 4, and interferon-γ, and inhibits lymphocyte proliferation (Klapproth et al., 1995, 1996, 2000; Malstrom and James, 1998). Similar activity is produced by other A/E pathogens, including EHEC O157:H7 strains and C. rodentium. The lifA gene is also known as Efa1 for EHEC factor for adherence as an Efa1 mutant in EHEC has significantly lower adherence to Chinese hamster ovary cells and is deficient in human red blood cell agglutination and autoaggregation (Nicholls et al., 2000). A similar phenotype is seen in EPEC, where the efa1 mutant is considerably less adherent to epithelial cells compared to its parent strain (Badea et al., 2003).
EspC
EspC is another large (110 kDa) secreted protein encoded on a pathogenicity island of EPEC (Vidal and Navarro-Garcia, 2008). EspC is a member of the SPATE (serine protease autotransporters of the enterobacteriaceae) family of autotransporter proteins encoding its own transport mechanism (see Chapter 16) (Henderson and Nataro, 2001). Such proteins possess: (a) an N-terminal signal sequence that promotes secretion through the inner membrane via the sec apparatus; (b) a C-terminal domain that forms a beta barrel pore in the outer membrane and exports; and (c) a central ‘passenger’ domain of the protein to the bacterial cell surface (Stein et al., 1996a). Although secreted via the type 5 mechanism, EspC requires the T3SS for translocation into host cells (Vidal and Navarro-Garcia, 2008). EspC causes cytotoxicity via cytoskeletal damage, which is dependent on the internalization of the serine protease motif (Navarro-Garcia et al., 2004). EspC is also an enterotoxin that induces a change in short-circuit current in rat jejunal tissue mounted on Ussing chambers (Mellies et al., 2001). The regulation of EspC is mediated by the global regulator Ler (Mellies et al., 1999).
Other toxins
EAEC strains produce an enterotoxin known as enteroaggregative heat-stable enterotoxin 1 (EAST1). A survey of diarrheagenic E. coli strains reported that 14 of 65 EPEC strains tested (22%) hybridized with an EAST1 probe (Savarino et al., 1996). The E2348/69 strain contains two copies of the east1 gene, one in the chromosome and another in the EAF plasmid.
Another toxin characterized in an EPEC strain is the cytolethal distending toxin (CDT) (Scott and Kaper, 1994). CDT is composed of three polypeptides cdtA, cdtB, and cdtC, where CDT-B is the catalytic subunit. The translocation of CDT-B is mediated by CDT-A and CDT-C subunits. Upon reaching the nucleus, CDT causes DNA damage (Ohara et al., 2004), i.e. chromatin disruption, leading to G2/M-phase growth arrest of the target cell and ultimately cell death (Lara-Tejero and Galan, 2000). There are other sporadic reports of production of CDT by EPEC. A study of CDT-producing E. coli in Bangladeshi children found that CDT-positive EPEC strains were isolated from more children with diarrhea than healthy controls; however, this difference did not reach statistical significance (Albert et al., 1995, 1996). Another study described the possible association of CDT-producing EPEC with diarrheal patients in the Republic of Korea (Kim et al., 2009). The E2348/69 strain does not encode CDT.
Other fimbriae and pili
Some EPEC strains elaborate pili or fimbriae other than or in addition to BFP (Bradley and Thompson, 1992; Girón et al., 1993). Atypical EPEC strains lacking BFP have been shown to express other fimbrial genes that could potentially aid in adherence to epithelial cells (Hernandes et al., 2011). The E. coli common pilus (ECP) has been implicated in the LA of aEPEC (Scaletsky et al., 2010b). The expression of the long, fine fimbriae; rigid bent fimbriae; and short, fine fimbriae is ablated in a ler mutant of the EPEC strain E2348/69 (Elliott et al., 2000).
Flagella have also been shown to be involved in EPEC adherence to epithelial cells. A mutation in the flagella gene, fliC, severely decreased the ability of EPEC to adhere to and form microcolonies on host cells. Adherence was also blocked by purified EPEC flagella and anti-flagellum antibodies (Girón et al., 2002). This ΔfliC phenotype was restored upon growth in preconditioned tissue culture media, suggesting that a signal from the mammalian cells influences flagella production.
Type I pili of EPEC have been found to be antigenic in volunteer studies (Karch et al., 1987). While type I pili do not have a role in adherence on epithelial cells in vitro (Elliott and Kaper, 1997), they have been associated with biofilm development at low temperatures in aEPEC O55:H7 (Weiss-Muszkat et al., 2010).
Adherence and invasion
Localized adherence
Typical EPEC strains adhere to HeLa, HEp-2, and other cell lines, and to organ cultures in vitro, in a distinctive pattern of three-dimensional clusters or microcolonies (Clausen and Christie, 1982; Scaletsky et al., 1984; Knutton et al., 1987a,b; Nataro et al., 1985, 1987; Yamamoto et al., 1992; Hicks et al., 1998). The LA pattern is a characteristic specific to EPEC strains and therefore has been used widely as a diagnostic tool (Figure 4.1A). A similar adherence pattern had been observed on tissue biopsies from EPEC-infected infants (Rothbaum et al., 1982, 1983). LA is due to the production of BFP by the bacterium. BFP binds to specific receptors on the host cell surface. Different investigators have found that LA can be inhibited by various sugar moieties, including galactose (Vanmaele and Armstrong, 1997), N-acetylgalactosamine (Scaletsky et al., 1988), N-acetyllactosamine (Vanmaele et al., 1999), and fucosylated oligosaccharides and gangliosides from milk (Jagannatha et al., 1991; Costa-Carvalho et al., 1994). These saccharides could be moieties of host cell glycolipids or glycoproteins that serve as EPEC receptors (Vanmaele et al., 1995; Manjarrez-Hernandez et al., 1997). A binding affinity between bundlin, the major structural subunit of BFP and N-acetyllactosamine has been established (Hyland et al., 2008). Although this interaction is weak, it may have a significant effect on LA due to the potentially large numbers of ligands and receptors involved. More recent work indicates that N-acetyllactosamine may trigger BFP retraction in addition to acting as a competitive inhibitor of EPEC binding to the host receptors (Hyland et al., 2006). EPEC also binds to phosphatidylethanolamine, a component of cell membranes (Foster et al., 1999; Barnett et al., 2000; Khursigara et al., 2001).
Other surface components contributing to EPEC adherence include intimin, the EspA filament and other fimbriae and/or pili. Single, double, and triple mutants of the bfp, espA, and eae genes revealed that while BFP and EspA both play a role in adherence, BFP plays a more dominant role; atypical EPEC strains that do not express BFP may use EspA for adherence. While a triple bfpA espAeae mutant is not adherent to epithelial cells (Cleary et al., 2004). EPEC strains lacking BFP remain able to adhere to pediatric small intestine tissue explants, although they form smaller colonies that lack the three-dimensional structure (Hicks et al., 1998). Initial adherence in such strains has been attributed to the production of E. coli common pilus (ECP) (Scaletsky et al., 2010a,b) and in some strains to the afimbrial adhesion, locus for diffuse adherence (LDA) (Torres et al., 2007). E. coli strains fitting the definition of EPEC but having non-LA pattern of adhesion, i.e. diffused adherence, have been noted (Knutton et al., 1991; Rodrigues et al., 1996; Beinke et al., 1998; Pelayo et al., 1999; Scaletsky et al., 1999). This phenotype has been attributed to the omicron subtype of intimin where the adherence was seen due to an invasive process (Hernandes et al., 2008).
Autoaggregation
When grown in tissue culture medium at 37°C, tEPEC strains aggregate into large clusters that may contain hundreds or thousands of bacteria (Figure 4.1B) (Vuopio-Varkila and Schoolnik, 1991). These autoaggregates are readily visible under a low-power microscope or even to the naked eye. They are unstable, dispersing quickly when moved to sub-optimal conditions (Bieber et al., 1998), with microcolonies in tissue culture forming and then dispersing over a period of 6 hours (Knutton et al., 1999). Like LA, autoaggregation requires BFP. Retraction of the pilus fiber causes bundlin pilin subunits to dissociate and thus the observed dispersion (Humphries et al., 2010). The BfpF protein drives pilus retraction and thus a bfpF mutant fails to disperse over time (Knutton et al., 1999).
Attaching and effacing
EPEC has the ability to strikingly alter the surface of the cells to which they attach. The characteristic phenotype observed during EPEC infection is the formation of A/E lesions (Moon et al., 1983), in which the brush border microvilli are sloughed off the apical surface of enterocytes (effacement) and transiently replaced by elongated microvillus-like processes (Frankel et al., 1996) (Figure 4.1C). Effacement of microvilli is followed by actin polymerization (Figure 4.1D) and ultimately the formation of prominent cup-like pedestals and elongated (up to 10 µm) pseudopod structures (Rosenshine et al., 1996a,b). EPEC utilize these pedestals to attach in very close (intimate) apposition to the host cell membrane. The pedestal structures are dynamic, being able to change length, shape, and position over time, and move the attached EPEC along the host cell surface (Sanger et al., 1996). A/E lesions are observed in model EPEC infections with cultured cells and mucosal explants (Knutton et al., 1987a,b; Hicks et al., 1998) as well as in intestinal biopsies from EPEC-infected infants or animals (Rothbaum et al., 1983; Peeters et al., 1988), but not on formalin-fixed epithelial cells (Knutton et al., 1997). The ability to form A/E lesions is shared among EPEC, EHEC, and strains of Escherichia albertii and C.rodentium (Tzipori et al., 1986; Schauer and Falkow, 1993; Huys et al., 2003). All attaching and effacing pathogens carry the LEE, which harbors the genes encoding intimin, eae, the adhesin required for attaching and effacing and the T3SS (Elliott et al., 1998).
Invasion
The ability of EPEC to be internalized by epithelial cells has been noted both in tissue culture (Andrade et al., 1989; Donnenberg et al., 1989, 1990b) and in small intestinal biopsies from EPEC-infected infants (Fagundes-Neto et al., 1995). Several critical EPEC virulence genes, including the bfp operon and the LEE, were first identified via the characterization of mutants lacking the ability to invade HEp-2 cells (Donnenberg et al., 1990a,b). The ability of EPEC to invade epithelial cells is in marked contrast to their capacity to evade phagocytic engulfment. Invasion requires the same virulence factors as A/E, therefore, it has been suggested that invasion is a byproduct of the cytoskeletal rearrangements that occur during the A/E process (Rosenshine et al., 1996b). MAP, or mitochondrial associated protein, is located immediately upstream of the operon encoding intimin and Tir. It is targeted to the host cell mitochondrial membrane where it disrupts the membrane potential. Map also mediates filopodia formation via Cdc42 (Kenny and Jepson, 2000; Kenny et al., 2002). This latter function of MAP has been shown to be involved in EPEC invasion (Jepson et al., 2003). An invasion phenotype has also been associated with other T3S effectors. EspT causes membrane ruffling and the generation of lamellipodia, via Rac1 and Cdc42 activation, leading to EPEC invasion (Bulgin et al., 2009). EspF and one of its host binding proteins, sorting nexin 9 (SNX9), which regulates vesicle trafficking and endocytosis, has also been shown to aid in the epithelial cell invasion of EPEC (Weflen et al., 2010). Despite its invasive potential, EPEC remains classified as an extracellular pathogen; this is likely due to the rarity of severe inflammation and bacteremia associated with EPEC disease.
Avoidance of host responses
Subversion by the T3SS
Like other bacterial pathogens, EPEC has multiple activators of host pattern recognition receptors including lipopolysaccharide, flagellin, and unmethylated CpG nucleotides and must contend with the host innate immune response to establish infection. Histologic examination of EPEC-infected tissue confirms the induction of an inflammatory response by these bacteria (Ulshen and Rollo, 1980; Higgins et al., 1999a,b; Marchès et al., 2000). However, some reports noted that the response is rather mild (Rothbaum et al., 1982). In cultured epithelial cells, EPEC activates NF-κB which induces the transcription of interleukin 8 (IL-8) (Figure 4.2C) (Savkovic et al., 1996, 1997). NF-κB functions as a homo- or heterodimer composed of various subtypes, e.g. RelA (p65), RelB, c-Rel, p50, and p52. This transcription factor is inactive while sequestered in the cytoplasm in an IκB-bound complex. Upon stimulation, IκB is phosphorylated by IκB kinase (IKK) and targeted for degradation, releasing NF-κB and allowing the subunits to translocate to the nucleus where they regulate gene expression (Karin and Ben-Neriah, 2000; Chen, 2005). EPEC also induces the activation of the MAP kinase cascade. c-Jun N-terminal kinases (JNKs) are serine/threonine kinases which belong to the MAP kinase family. Upon activation, JNKs phosphorylate c-Jun, an AP-1 transcription factor. Despite measurable activation of these pro-inflammatory cascades, it appears that multiple EPEC T3S effectors also negatively regulate the host cell response,with T3SS mutants inducing greater levels of activation (Sham et al., 2011). Most of these effectors are interdependent, multifunctional and redundant (Dean and Kenny, 2009). More details on T3SS effectors can be found in Chapter 15.
The E2348/69 strain contains two nleH genes, both of which have been shown to inhibit pro-inflammatory cytokine expression and promote colonization (Royan et al., 2010). Both NleH1 and NleH2 reduce the abundance of nuclear ribosomal protein S3 (RPS3), a non-Rel subunit of p65 homodimer and p65–p50 heterodimer (Gao et al., 2009), however, only NleH1 inhibited the NF-κB activity independent of IκBα phosphorylation and degradation (Pham et al., 2012). NleC and NleD are zinc metalloproteases that specifically cleave NF-κBp65 and JNK respectively (Baruch et al., 2011). This cleavage prevents transcription of NF-κB- and AP-1-induced genes leading to a marked reduction in IL-8 secretion (Yen et al., 2010; Baruch et al., 2011). NleC has also been found to bind and cleave host acetyltransferase p300, decreasing the inflammatory response. NleB and NleE act to inhibit p65 translocation to the nucleus (Newton et al., 2010). NleB inhibits the TNF-α pathway, while NleE is an S-adenosyl-L-methionine-dependent methyltransferase that prevents the activation of IKKβ (Nadler et al., 2010) by inactivating its kinase activity (Zhang et al., 2012). Recently, Tir has been shown to inhibit NF-κB activation by inducing TNF-α receptor associated factor (TRAF) protein degradation (Ruchaud-Sparagano et al., 2011).
Apoptosis is another part of the host innate immune response induced by bacterial infection; this action aids in the elimination of the infecting pathogen and promotes a dendritic-cell-mediated immune response. EPEC subverts this response at multiple stages throughout the process. UV-induced apoptosis is JNK-dependent and may be inhibited by transient expression of NleD. In addition, infection with an nleD mutant strain of EPEC induces a greater percentage of apoptosis compared with wild-type. These results indicate that NleD inhibits JNK-dependent apoptosis, likely via JNK cleavage (Baruch et al., 2011). EspZ, a LEE-encoded effector, promotes the survival of epithelial cells by enhancing the phosphorylation of the focal adhesion kinase (FAK) and thereby may stabilize the epithelium upon infection (Shames et al., 2010). NleH inhibits apoptosis by preventing pro-caspase-3 cleavage through direct binding to Bax inhibitor-1 (BI-1) protein (Hemrajani et al., 2010). The NleF effector, highly conserved in EPEC and EPEC, binds the active site of caspase-9 to inhibit its activity (Blasche et al., 2013). For additional information, see ‘Apoptosis’, below.
Damage
Cytoskeletal rearrangement
Numerous signaling and actin-associated/binding proteins including α-actinin, ezrin, and myosin light chain II (MLC) are recruited to the site of bacterial intimate adhesion (Manjarrez-Hernandez et al., 1996; Cantarelli et al., 2001; Goosney et al., 2001). These proteins are involved in pedestal formation, which leads to loss of absorptive surface (effacing). EPEC achieves this effect by subverting fundamental host cell functions to build focal adhesion-like structures that anchor the bacteria to the host cytoskeleton. Upon EPEC infection and insertion of Tir into the host cell membrane, Tir becomes clustered at the site of bacterial attachment, where it serves as the receptor for intimin. F-actin and other host cytoskeletal proteins accumulate at this site and generate a pedestal (Figure 4.2B) (Knutton et al., 1987a,b; da Silva et al., 1989; Finlay et al., 1992). The localized F-actin accumulation can be demonstrated using fluorescent probes, which is the basis of the diagnostic fluorescent-actin staining (FAS) test (Figure 4.1D). Recent advances have begun to decipher the detailed molecular architecture of the pedestals and how EPEC remodels the host cytoskeleton at the surface of the cell. These studies have revealed that different strains of EPEC use different pathways to achieve the same end. For the E2348/69 strain, Tir is phosphorylated on Y474 by redundant kinases from the Src family, including Fyn and Tec/Abl family kinases (Phillips et al., 2004; Bommarius et al., 2007). This phosphorylation is essential for actin remodeling and pedestal formation (Rosenshine et al., 1996a,b; Kenny, 1999) but it is not required for the membrane insertion of Tir or intimin binding (Kenny et al., 1997; Gauthier et al., 2000). After phosphorylation, the residues flanking Y474 directly bind to the host SH3/SH2 adaptor protein Nck (Gruenheid et al., 2001; Campellone et al., 2002). Nck recruits the neural-Wiskott-Aldrich syndrome protein (N-WASP), which activates the actin-related proteins 2 and 3 (Arp2/3) complex and leads to the formation of the actin pedestals (Figure 4.2B) (Lommel et al., 2001). However, some EPEC strains lacking Tir Y474 are still capable of promoting actin polymerization. These Nck-independent modes of recruiting N-WASP are attributed to phosphorylation of Y454 (Campellone and Leong, 2005), and to the TccP (Tir-cytoskeleton coupling protein) (Whale et al., 2006) effectors one and two (also known as EspFU and EspFM). These effectors bind the GTPase binding domain of N-WASP (Campellone et al., 2004). More recently, phosphorylation of Y474 has been shown to recruit redundant kinases. The polyproline region of EspF interacts with the SH3 domains of these kinases, while the Y474 site interacts with the SH2 domains (Bommarius et al., 2007). This activity constitutes a positive feedback loop in which the recruited kinases phosphorylate other Tir molecules.
Recent studies have highlighted the importance of other T3S effectors in remodeling the host cytoskeleton by modulating Rho GTPases. EspF is a 206-amino-acid protein, the C-terminus of which contains three identical repeats of a proline-rich sequence that resembles those recognized by eukaryotic signaling proteins containing SH3 domains (Donnenberg et al., 1997). The C-terminus of each repeat also contains an N-WASP binding motif (Alto et al., 2007). EspF activates the Arp2/3 complex and induces actin polymerization in vitro (Weflen et al., 2009). Map and EspM display guanine exchange factor (GEF) activity (Huang et al., 2009; Arbeloa et al., 2010). Map and EspT activate Cdc42 activity, which induces filopodia formation (Kenny et al., 2002; Berger et al., 2009), while EspM activates RhoA, inducing actin stress fibers (Alto et al., 2006; Arbeloa et al., 2008). Other T3S effectors involved in pedestal dynamics are EspH and EspV (Tu et al., 2003; Arbeloa et al., 2011). The mechanism by which EspV modulates the cytoskeleton is unknown. EspH competitively binds to tandem DH-PH domains of endogenous Dbl-homology and pleckstrin-homology RhoGEFs and prevents Rho activation (Dong et al., 2010). Thus, EPEC strains secrete effectors that inactivate endogenous RhoGEFs, in addition to secreting their own GEF to hijack the host signaling pathway. EspH also stimulates actin polymerization by recruiting N-WASP and the Arp2/3 complex independently of the tyrosine phosphorylation of Tir but via the C-terminus of Tir and the WH1 domain of N-WASP (Wong et al., 2012a,b).
Disruption of the epithelial barrier
The apical junction complex, which consists of tight junctions (TJ) and adherent junctions connecting neighboring cells, is critical for separation of intestinal tissues from luminal contents (Harhaj and Antonetti, 2004). The TJ is comprised of three different transmembrane proteins; occludin, claudins, and junctional adhesion molecule (JAM). These proteins are attached to actin filaments and MLC via the zonula occludens (ZO) family of proteins, which are peripheral membrane proteins associated with the cytoplasmic surface of TJ and adherent junctions. Adherent junctions are composed of the transmembrane protein E-cadherin and cytoplasmic proteins from the catenin family (Kuphal and Bosserhoff, 2012).
EPEC infection of polarized cultured intestinal cells (Caco-2 or T84) increases the permeability of the monolayer, as reflected by a decrease in transepithelial electrical resistance (TER) and a flux of markers across the monolayer via the paracellular pathway. These permeability changes are mediated by the modification of the TJ structure (Canil et al., 1993; Spitz et al., 1995; Philpott et al., 1996). EPEC infection activates the MLC kinase which phosphorylates MLC, inducing the contraction of the actinomysin ring and increasing paracellular permeability (Manjarrez-Hernandez et al., 1996). EPEC also induces the redistribution of occludin, ZO-1, clearing of claudin-1 (Simonovic et al., 2000; McNamara et al., 2001; Muza-Moons et al., 2004; Shifflett et al., 2005) and relocalization and activation of ezrin, a protein that links TJ proteins to the actin cytoskeleton (Simonovic et al., 2001). This redistribution decreases protein interactions at the TJ and disrupts barrier function. Redistribution of these junctional proteins is dependent on the EspB translocation protein and on the effector EspF (Simonovic et al., 2001). EspB is also required for the phosphorylation of protein kinase C which increases association with E-cadherins resulting in redistribution of β-catenin and increased paracellular permeability (Malladi et al., 2004). EPEC infection causes β1-integrins to migrate from the basolateral side to the apical side, where they interact with intimin and enhance barrier defects (Muza-Moons et al., 2003). Another T3S effector, EspG, alters paracellular permeability but does not alter the transepithelial electrical resistance (Matsuzawa et al., 2005). EspG also causes localized depletion of microtubules triggering disruption in the network (Shaw et al., 2005).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


