As a reasonable generalization the microvascular complications (retinopathy, nephropathy, and peripheral neuropathy) are markedly influenced by good blood glucose control, while the macrovascular complications (myocardial infarction, stroke, and peripheral vascular disease) are impacted much more by blood pressure control and much less by tight glucose control.
The pathogenesis of the micro- and macrovascular complications of diabetes are distinct: microvascular changes reflect the impact of high glucose concentrations on biochemical processes (advanced glycosylation end products, sorbitol accumulation, for example, as well as increased glomerular filtration); macrovascular complications represent accelerated atherosclerotic changes related to standard cardiovascular risk factors (blood pressure, lipids). The differences in pathogenetic mechanisms account for the differences in response to glucose lowering.
 Microvascular complications are much more common in patients with type 1 diabetes than in patients with type 2; since type 2 is five times more common than type 1, however, there are more type 2 patients with microvascular complications in the population at large.
Microvascular complications are much more common in patients with type 1 diabetes than in patients with type 2; since type 2 is five times more common than type 1, however, there are more type 2 patients with microvascular complications in the population at large.
 In type 1 diabetics the microvascular complications are related to the length of time that diabetes has been present; in type 2 patients microvascular complications may be present at the initial presentation.
In type 1 diabetics the microvascular complications are related to the length of time that diabetes has been present; in type 2 patients microvascular complications may be present at the initial presentation.
This likely reflects the fact that the onset of type 1 diabetes is generally known with precision: a dramatic onset usually with ketoacidosis. In type 2 diabetes the onset is insidious rather than dramatic, and abnormal glucose metabolism is often present for years before diagnosis.
 The cause of diabetes is always insulin deficiency but there is an important difference between type 1 and type 2: in type 1 the deficiency of insulin is absolute and complete; in type 2 the insulin deficiency is relative to the needs imposed by insulin resistance.
The cause of diabetes is always insulin deficiency but there is an important difference between type 1 and type 2: in type 1 the deficiency of insulin is absolute and complete; in type 2 the insulin deficiency is relative to the needs imposed by insulin resistance.
In type 2 the insulin levels are elevated compared with nondiabetic subjects but not elevated enough to lower the glucose levels to normal. Eventually, in many type 2 patients beta cell function slowly declines, necessitating treatment with insulin.
Diabetic Ketoacidosis
 Diabetic ketoacidosis (DKA) develops in the complete absence of insulin, usually in the setting of type 1 diabetes mellitus (Table 7-1).
Diabetic ketoacidosis (DKA) develops in the complete absence of insulin, usually in the setting of type 1 diabetes mellitus (Table 7-1).
Under these circumstances lipolysis is totally unrestrained, free fatty acids are converted in the liver to ketone bodies (beta-hydroxybutyrate and oxaloacetate). Bedside testing with diluted serum and an Acetest tablet is sufficient to make the diagnosis in a patient with an anion gap.
 Ketoacidosis never occurs in simple fasting since the amount of insulin available during a fast, although low, is not absent, and remains sufficient to prevent the unrestrained lipolysis characteristic of DKA.
Ketoacidosis never occurs in simple fasting since the amount of insulin available during a fast, although low, is not absent, and remains sufficient to prevent the unrestrained lipolysis characteristic of DKA.
TABLE 7.1 Diabetic Comas

Simple fasting is associated with ketosis but the latter does not produce enough ketoacids to cause a metabolic acidosis. During fasting or starvation urine ketones are positive but serum ketones are absent or present in very low concentration.
 The commonest cause of DKA is failure to take insulin.
The commonest cause of DKA is failure to take insulin.
DKA frequently occurs in the setting of some (frequently mild) intercurrent illness in insulin-dependent diabetics; the patient, fearing that he or she will not be able to eat, omits insulin to avoid hypoglycemia. The intercurrent illness, however, increases insulin resistance and accentuates the need for insulin. More, not less, insulin is needed in these circumstances. This is an educational issue; patients should be instructed not to discontinue insulin when they become ill, but to follow “sick day rules” and contact their physician for instruction.
 DKA is associated with significant potassium depletion. The initial serum potassium level may be elevated (masking the deficit) because acidosis is associated with intracellular shifts of hydrogen ions and potassium.
DKA is associated with significant potassium depletion. The initial serum potassium level may be elevated (masking the deficit) because acidosis is associated with intracellular shifts of hydrogen ions and potassium.
The acidosis is buffered in part by cellular uptake of H+ which is associated with potassium efflux into the extracellular space. As the acidosis diminishes during treatment the intracellular shifts are reversed and potassium levels fall. Potassium must be closely monitored and administered intravenously as the levels fall. A “normal” potassium level at presentation with acidosis necessitates potassium replacement from the outset.
 The EKG is a convenient way to assess potassium status at the bedside in DKA; initially high extracellular levels are reflected in high spiked T waves.
The EKG is a convenient way to assess potassium status at the bedside in DKA; initially high extracellular levels are reflected in high spiked T waves.
As the potassium level falls the T waves flatten indicating the need for replacement of potassium. Potassium levels should be measured hourly along with glucose, electrolytes, and the anion gap for as long as the ketoacidosis persists.
 The issues to be addressed in DKA are volume depletion, acidosis, hyperosmolality, hypoglycemia, and serum potassium levels.
The issues to be addressed in DKA are volume depletion, acidosis, hyperosmolality, hypoglycemia, and serum potassium levels.
Patients with DKA are severely volume depleted and dehydrated at presentation because of the osmotic diuresis produced by glycosuria, poor fluid intake, and occasionally, vomiting.
 The osmotic diuresis from uncontrolled glucosuria produces losses of water and salt (the urine osmolality during osmotic diuresis is about half normal saline, so more water than salt is lost).
The osmotic diuresis from uncontrolled glucosuria produces losses of water and salt (the urine osmolality during osmotic diuresis is about half normal saline, so more water than salt is lost).
This should be addressed by aggressive fluid resuscitation; normal saline can be given immediately, but a quick calculation of the osmolality (followed by a direct measurement in the laboratory) should be made and the infusate modified accordingly. Most patients will be hyperosmolar and require dilute fluids (half normal saline).
 A serum osmolality of 340 mOsm/kg or greater is an emergency that requires prompt treatment with hypotonic fluids.
A serum osmolality of 340 mOsm/kg or greater is an emergency that requires prompt treatment with hypotonic fluids.
Diffuse cerebral bleeding from brain shrinkage as the cortex pulls away from the meninges is the feared complication of hyperosmolar states.
 The primary treatment of the acidosis in DKA is to stop the production of ketoacids by administering insulin. Insulin stops the flux of free fatty acids (FFA) to the liver and promotes lipid, rather than ketoacid, synthesis.
The primary treatment of the acidosis in DKA is to stop the production of ketoacids by administering insulin. Insulin stops the flux of free fatty acids (FFA) to the liver and promotes lipid, rather than ketoacid, synthesis.
Insulin should be administered by a bolus followed by continuous infusion. It is important to avoid hypoglycemia so when the plasma glucose falls into the range of 250 to 300 mg/dL, glucose (5%) should be added to the IV fluids. Unless the acidosis is very severe (pH below 7) and the blood pressure fails to respond to fluids, bicarbonate administration should be avoided.
There are several very good reasons to avoid bicarbonate in the treatment of DKA:
1. It is sometimes associated with cerebral edema. The mechanisms leading to this catastrophic complication are obscure but may involve oxygen delivery to the brain. In DKA red cell 2,3-diphoshoglycerate (2,3-DPG) is diminished due to the acidosis and perhaps to diabetes. 2,3-DPG is an important regulator of tissue oxygen delivery by changing the affinity of hemoglobin for oxygen: high levels of 2,3-DPG promote oxygen release, while low levels decrease oxygen delivery. In DKA the adverse effect of low 2,3-DPG is compensated by the independent effect of acidosis which decreases hemoglobin oxygen affinity, thereby promoting oxygen release. Bicarbonate administration, by rapidly changing the pH, leaves the adverse effect of low 2,3-DPG unopposed and decreased oxygen delivery a possible result (see Fig 2.2). Whether this is responsible for the cerebral edema is not firmly established but the association of bicarbonate administration with the latter is well recognized.
2. Bicarbonate induces a swift fall in serum potassium levels. As described above acidosis induces a shift in potassium from the intracellular to the extracellular space so the initial level is frequently elevated masking severe potassium depletion; correcting the acidosis rapidly with bicarbonate leads to a rapid fall in potassium as cellular uptake of potassium increases; this shift may be associated with serious, sometimes fatal, arrhythmias. Accentuating hypokalemia during treatment is a serious problem.
3. Endogenous bicarbonate is generated from glucose metabolism during successful treatment of DKA. A metabolic alkalosis may develop if large amounts of bicarbonate have been administered in addition to insulin.
Hyperosmolar Nonketotic Coma
 Hyperosmolar nonketotic coma (HONC) is a syndrome that usually occurs in noninsulin-dependent (usually type 2) diabetics (Table 7-1).
Hyperosmolar nonketotic coma (HONC) is a syndrome that usually occurs in noninsulin-dependent (usually type 2) diabetics (Table 7-1).
In distinction to type 1 patients those at risk for hyperosmolar coma are not devoid of insulin. Since the effects of insulin on inhibiting lipolysis are more sensitive than those stimulating muscle glucose uptake, these type 2 patients do not have unrestrained lipolysis and do not develop ketoacidosis. They suffer, rather, the effects of very high glucose levels. The associated volume depletion is usually severe and frequently associated with hyperosmolality.
 Renal insufficiency is usually present allowing the glucose to rise to extremely high levels.
Renal insufficiency is usually present allowing the glucose to rise to extremely high levels.
The osmotic diuresis results in water and salt loss with the greater deficit in water leading to the hyperosmolar state. Treatment is fluid resuscitation with hypotonic fluids and insulin. Glucose should be added to the fluids when the plasma glucose level reaches 300 mg/dL.
 Intercurrent infections which increase insulin resistance and decrease fluid intake or treatment with glucocorticoids are common initiating events in HONC.
Intercurrent infections which increase insulin resistance and decrease fluid intake or treatment with glucocorticoids are common initiating events in HONC.
The mortality rate is high.
HYPOGLYCEMIA
Pathophysiology
Since the brain, for practical purposes, can utilize only glucose as an energy source hypoglycemia is completely analogous to hypoxia in its effects on neuronal function.
 In the postabsorptive and fasting state blood glucose is maintained by glycogenolysis and gluconeogenesis predominantly within the liver.
In the postabsorptive and fasting state blood glucose is maintained by glycogenolysis and gluconeogenesis predominantly within the liver.
The liver is almost completely depleted of glycogen after an overnight fast leaving gluconeogenesis as the prime defender of the blood glucose level in more prolonged fasts.
 In advanced liver failure hypoglycemia may occur, but this is usually a preterminal event.
In advanced liver failure hypoglycemia may occur, but this is usually a preterminal event.
 Spontaneous (endogenous) hypoglycemia occurs in either the fasting or the postprandial state. This is a clinically important distinction since the relationship of hypoglycemia to meals is an important clue to diagnosis (Table 7-2).
Spontaneous (endogenous) hypoglycemia occurs in either the fasting or the postprandial state. This is a clinically important distinction since the relationship of hypoglycemia to meals is an important clue to diagnosis (Table 7-2).
In both fasting and postprandial hypoglycemia the level of insulin is inappropriately high for the level of glucose.
Glucose Counterregulation
 Hypoglycemia calls forth a counterregulatory hormonal response which increases the plasma glucose level. Glucagon and epinephrine are the principal counterregulatory hormones.
Hypoglycemia calls forth a counterregulatory hormonal response which increases the plasma glucose level. Glucagon and epinephrine are the principal counterregulatory hormones.
Increases in the secretion of these hormones, along with suppression of endogenous insulin release, raise the blood glucose level by stimulating hepatic glucose output (glycogenolysis and gluconeogenesis). Epinephrine contributes additionally by providing substrate (lactate) for gluconeogenesis from glycogenolysis in muscle, and by providing alternative substrate for nonneural tissues by stimulating lipolysis in adipose tissue.
 The plasma glucose threshold for the initiation of the counterregulatory response is between 50 and 60 mg/dL.
The plasma glucose threshold for the initiation of the counterregulatory response is between 50 and 60 mg/dL.
Although cortisol and growth hormone (GH) are increased by hypoglycemia, neither of these hormones is involved in the acute counterregulatory response. Notwithstanding, the response of the pituitary to the intentional induction of hypoglycemia by intravenous insulin administration provides a convenient test of ACTH and GH reserve in the evaluation of pituitary function.
Symptoms of Hypoglycemia
 Symptoms of hypoglycemia are of two types: adrenergic and neuroglycopenic.
Symptoms of hypoglycemia are of two types: adrenergic and neuroglycopenic.
 Epinephrine release from the adrenal medulla as part of the counter regulatory response causes the adrenergic symptoms including palpitations and tremor along with the associated signs of tachycardia, widened pulse pressure, and increased systolic BP.
Epinephrine release from the adrenal medulla as part of the counter regulatory response causes the adrenergic symptoms including palpitations and tremor along with the associated signs of tachycardia, widened pulse pressure, and increased systolic BP.
TABLE 7.2 Hypoglycemia
These epinephrine-induced symptoms form an early warning system for hypoglycemia. Sweating is prominent but not, as noted below, a consequence of epinephrine.
 Impaired neuronal function is the manifestation of neuroglycopenia which includes confusion, dysarthria, impaired cognition, and in the extreme, seizures, coma, and even death.
Impaired neuronal function is the manifestation of neuroglycopenia which includes confusion, dysarthria, impaired cognition, and in the extreme, seizures, coma, and even death.
 Sweating, a very common manifestation of hypoglycemia, is cholinergically mediated eccrine sweating. It is due to the fall in temperature that occurs during hypoglycemia. Hypoglycemia is associated with mild hypothermia. Sweating dissipates heat to meet the new lower core temperature set point associated with hypoglycemia.
Sweating, a very common manifestation of hypoglycemia, is cholinergically mediated eccrine sweating. It is due to the fall in temperature that occurs during hypoglycemia. Hypoglycemia is associated with mild hypothermia. Sweating dissipates heat to meet the new lower core temperature set point associated with hypoglycemia.
Patients may be drenched in sweat particularly during severe episodes. It is a faux pearl that the sweating is adrenergically mediated. Adrenergic sweating is apocrine in type and involves the axillary sweat glands and those about the upper lip. Hypoglycemic diaphoresis is generalized and involves the cholinergic sympathetic nerves.
 Whipple’s triad, the classic if self-evident criteria for the diagnosis of hypoglycemia, consists of: 1) compatible symptoms, 2) low measured glucose during the symptoms, and 3) relief with glucose administration.
Whipple’s triad, the classic if self-evident criteria for the diagnosis of hypoglycemia, consists of: 1) compatible symptoms, 2) low measured glucose during the symptoms, and 3) relief with glucose administration.
The symptoms of hypoglycemia, particularly the adrenergic symptoms, are relatively nonspecific and mimic those caused by anxiety. A normal glucose during an episode categorically rules out hypoglycemia and thus the need for further evaluation. Most patients who believe that they have hypoglycemia do not fulfil Whipple’s triad.
Causes of Hypoglycemia
 Drugs (insulin, sulfonylureas) are important causes of hypoglycemia.
Drugs (insulin, sulfonylureas) are important causes of hypoglycemia.
 Exogenous insulin administration, by far the most common cause of hypoglycemia, is easily distinguished from endogenous hyperinsulinism by the absence of C peptide in the blood.
Exogenous insulin administration, by far the most common cause of hypoglycemia, is easily distinguished from endogenous hyperinsulinism by the absence of C peptide in the blood.
C peptide is the connecting piece of the insulin molecule that is cleaved during insulin synthesis and that is absent in commercial insulin preparations. It is released into the circulation along with insulin from the pancreatic beta cells.
 Insulin-induced hypoglycemia is usually a consequence of treatment for diabetes gone awry.
Insulin-induced hypoglycemia is usually a consequence of treatment for diabetes gone awry.
It is particularly common when the most intensive insulin regimens are employed.
 Insulin-induced hypoglycemia may also occur in suicide attempts or as a manifestation of factitious disease.
Insulin-induced hypoglycemia may also occur in suicide attempts or as a manifestation of factitious disease.
The latter should be suspected when hypoglycemia is noted in hospital workers or in the family of patients taking insulin.
 The physiologic side effects of epinephrine (tachycardia, palpitations, tremor), along with sympathetic cholinergic sweating, provide early warning signs of hypoglycemia.
The physiologic side effects of epinephrine (tachycardia, palpitations, tremor), along with sympathetic cholinergic sweating, provide early warning signs of hypoglycemia.
When the epinephrine response is deficient, as in certain forms of autonomic failure, neuroglycopenia catches the patient unaware and may be associated with serious clinical manifestations such as seizures and coma.
 Hypoglycemia unawareness is a significant problem in long-term insulin-dependent diabetics and underlies the occurrence of repeated hypoglycemic episodes in these patients.
Hypoglycemia unawareness is a significant problem in long-term insulin-dependent diabetics and underlies the occurrence of repeated hypoglycemic episodes in these patients.
Failure of the glucagon response is common in long-standing insulin-dependent diabetics with absolute insulin deficiency. Insulin within the islets appears necessary to maintain glucagon responsiveness. To mount a counterregulatory response these diabetic patients are critically dependent on epinephrine; should autonomic failure develop the risk of repeated episodes of hypoglycemia is markedly increased.
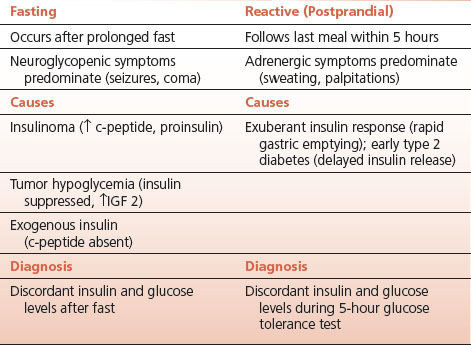
 Fasting hypoglycemia occurs in the postabsorptive state well after the last meal. Insulinoma is the most common cause. Neuroglycopenic symptoms tend to dominate the clinical course.
Fasting hypoglycemia occurs in the postabsorptive state well after the last meal. Insulinoma is the most common cause. Neuroglycopenic symptoms tend to dominate the clinical course.
 The diagnosis of insulinoma is established by the discordance of glucose and insulin levels in the fasting state.
The diagnosis of insulinoma is established by the discordance of glucose and insulin levels in the fasting state.
This can be demonstrated by obtaining serial morning fasting plasma for glucose and insulin, or by a 72-hour fast. Measurements of C peptide level and proinsulin in plasma are also of value as these precursors of the mature insulin molecule may be elevated in patients with insulinoma.
 Postprandial or “reactive” hypoglycemia occurs from 1 to 5 hours after a meal depending on the underlying disorder. The diagnosis can usually be established by a 5-hour glucose tolerance test with simultaneous measurements of insulin.
Postprandial or “reactive” hypoglycemia occurs from 1 to 5 hours after a meal depending on the underlying disorder. The diagnosis can usually be established by a 5-hour glucose tolerance test with simultaneous measurements of insulin.
Higher than normal levels of insulin in response to the meal are presumed to be the cause, so that discordance between insulin and glucose levels occurs as the postprandial rise in glucose wanes while insulin is still present in sufficient quantities to lower the glucose into hypoglycemic ranges. Neuroglycopenic symptoms are very rare in reactive hypoglycemia, although they do occasionally occur. Hypoglycemia within an hour or two of a meal may occur in patients who have had gastric surgery (tachyalimentation) with rapid gastric emptying and a concomitant excessive rise in insulin. Hypoglycemia at 5 hours may be seen in early diabetes since the insulin response is delayed in these patients.
 Fasting hypoglycemia may also be caused by nonbeta-cell tumors: tumor hypoglycemia.
Fasting hypoglycemia may also be caused by nonbeta-cell tumors: tumor hypoglycemia.
Originally described with large, bulky, mesenchymal tumors the hypoglycemia was thought to result from glucose utilization by the tumor. This was never a very compelling explanation since hepatic glucose output was suppressed by the tumor and muscle glucose uptake was enhanced. Insulin production by the nonbeta-cell tumors was excluded as a cause, prompting a search for other humoral tumor products that might be responsible for these insulin-like effects.
 Production of insulin-like growth factor 2 (IGF2) by the tumor is the cause of tumor hypoglycemia in the majority of cases. The hypoglycemia is frequently severe and may be the presenting manifestation of the tumor.
Production of insulin-like growth factor 2 (IGF2) by the tumor is the cause of tumor hypoglycemia in the majority of cases. The hypoglycemia is frequently severe and may be the presenting manifestation of the tumor.
Incompletely processed IGF2 stimulates insulin receptors with resultant hypoglycemia. Insulin itself is suppressed. Many different types of tumor have been associated with this rare cause of hypoglycemia. Removing the tumor, wherever possible, is the treatment.
 Alcoholic hypoglycemia occurs when ethanol is ingested in the starved state and maintenance of the plasma glucose is critically dependent on gluconeogenesis. The metabolism of the ethanol floods the liver with NADH, thereby blocking the synthesis of glucose from gluconeogenic precursors and disrupting the fasting equilibrium that maintained the plasma glucose by gluconeogenesis.
Alcoholic hypoglycemia occurs when ethanol is ingested in the starved state and maintenance of the plasma glucose is critically dependent on gluconeogenesis. The metabolism of the ethanol floods the liver with NADH, thereby blocking the synthesis of glucose from gluconeogenic precursors and disrupting the fasting equilibrium that maintained the plasma glucose by gluconeogenesis.
The typical picture of alcoholic hypoglycemia involves a serious alcoholic binge followed by a period of stupor. Fuel reserves are typically depleted by the preceding drunken spree and unconscious interval. On awakening alcohol consumption is resumed and the first drink initiates the biochemical cascade that blocks gluconeogenesis and hypoglycemia is the result.
MULTIPLE ENDOCRINE NEOPLASIA SYNDROMES
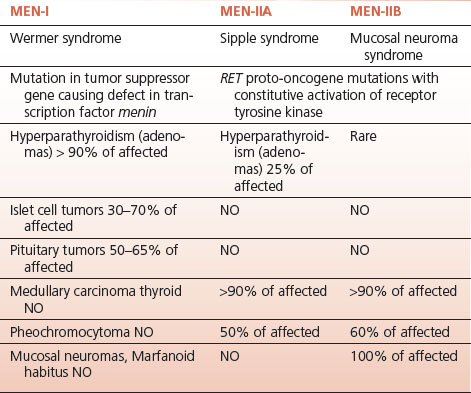
 The multiple endocrine neoplasia (MEN) syndromes comprise three genetically and clinically distinct familial entities that result in adenomatous hyperplasia and malignant tumor formation in different endocrine organs (Table 7-3).
The multiple endocrine neoplasia (MEN) syndromes comprise three genetically and clinically distinct familial entities that result in adenomatous hyperplasia and malignant tumor formation in different endocrine organs (Table 7-3).
The clinical manifestations depend upon the pattern of glandular involvement and hormone secretion. All three have autosomal dominant inheritance with high penetration and variable expressivity. The genes involved have been identified and genetic screening is available for each syndrome.
MEN 1
 MEN 1 involves the pancreas, the pituitary, and the parathyroid glands (the three Ps.).
MEN 1 involves the pancreas, the pituitary, and the parathyroid glands (the three Ps.).
TABLE 7.3 Multiple Endocrine Neoplasia (MEN) Syndromes
Any combination of tumors is possible; an affected individual with one of the tumors has a lifelong risk for the development of tumors in the other glands, necessitating lifelong screening. About 40% of affected persons develop tumors in all three glands. MEN 1 is caused by a mutation in a tumor suppressor gene that encodes a transcription factor called “menin.”
 Parathyroid tumors causing hypercalcemia are the most common manifestation of MEN 1 occurring in over 90% of affected individuals.
Parathyroid tumors causing hypercalcemia are the most common manifestation of MEN 1 occurring in over 90% of affected individuals.
Although asymptomatic hypercalcemia is most common, nephrolithiasis and nephrocalcinosis may occur. Diffuse parathyroid hyperplasia or multiple adenomas are more common than solitary adenomas, unlike the sporadic form of hyperparathyroidism in which a solitary adenoma predominates.
 Pancreatic islet cell tumors occur in 60% to 70% of those affected with MEN 1; they may secrete insulin, gastrin, or vasoactive intestinal peptide (VIP) giving rise to syndromes based on hypersecretion of these hormones.
Pancreatic islet cell tumors occur in 60% to 70% of those affected with MEN 1; they may secrete insulin, gastrin, or vasoactive intestinal peptide (VIP) giving rise to syndromes based on hypersecretion of these hormones.
Most tumors secrete pancreatic polypeptide as well, although the latter has no known clinical sequelae. Diffuse hyperplasia or multiple adenomas are the rule, and tumors may occur in the duodenal wall as well as in the pancreas.
 Insulinomas originating from islet beta cells comprise about 40% of pancreatic tumors in MEN 1; they cause fasting hypoglycemia.
Insulinomas originating from islet beta cells comprise about 40% of pancreatic tumors in MEN 1; they cause fasting hypoglycemia.
Only a minority of patients with insulinoma, however, have the MEN 1 syndrome.
 Gastrinomas originate from nonbeta-cell elements of the pancreas or the duodenal wall. They cause severe gastric acid overproduction (Zollinger–Ellison syndrome) which is associated with severe atypical peptic ulcer disease.
Gastrinomas originate from nonbeta-cell elements of the pancreas or the duodenal wall. They cause severe gastric acid overproduction (Zollinger–Ellison syndrome) which is associated with severe atypical peptic ulcer disease.
About 50% of patients with gastrinomas have the MEN 1 syndrome.
 Less common nonbeta-cell tumors secrete VIP (VIPomas) causing the syndrome of watery diarrhea, hypokalemia, and achlorhydria (WDHA, pancreatic cholera, Verner Morrison syndrome).
Less common nonbeta-cell tumors secrete VIP (VIPomas) causing the syndrome of watery diarrhea, hypokalemia, and achlorhydria (WDHA, pancreatic cholera, Verner Morrison syndrome).
Nonbeta-cell islet tumors in MEN 1 may also rarely secrete ACTH (causing Cushing’s syndrome), calcitonin, and glucagon.
Carcinoid tumors of embryologic foregut origin giving rise to an atypical carcinoid syndrome is rarely associated as well.
 Pituitary tumors in MEN 1 occur in less than 50% of affected patients; they may produce prolactin, GH, and very rarely, ACTH.
Pituitary tumors in MEN 1 occur in less than 50% of affected patients; they may produce prolactin, GH, and very rarely, ACTH.
Galactorrhea, infertility, impotence, acromegaly and, rarely, Cushing’s disease may occur depending on the secretion products of the tumor.
MEN 2A
 MEN 2A consists of medullary thyroid carcinoma (MCT) (>90% of affected individuals), pheochromocytoma (about 50% of those affected), and hyperparathyroidism (<20% of those with MEN 2A).
MEN 2A consists of medullary thyroid carcinoma (MCT) (>90% of affected individuals), pheochromocytoma (about 50% of those affected), and hyperparathyroidism (<20% of those with MEN 2A).
The genetic abnormality is well characterized (chromosome 10) and consists of mutations in the RET proto-oncogene that produces a constitutive activation in a receptor tyrosine kinase. All family members should be screened for this mutation.
Pheochromocytoma is the short-term immediate problem. Most kindreds have been identified by a symptomatic pheochromocytoma in the proband.
 The pheochromocytomas in MEN 2A are always intra-adrenal, usually multicentric within the adrenal, preceded by adrenal medullary hyperplasia, frequently bilateral at presentation, and always epinephrine secreting.
The pheochromocytomas in MEN 2A are always intra-adrenal, usually multicentric within the adrenal, preceded by adrenal medullary hyperplasia, frequently bilateral at presentation, and always epinephrine secreting.
Pheochromocytoma should be ruled out (measurement of urinary epinephrine and metanephrine) and, if present, addressed surgically before operation on the thyroid or parathyroids. If imaging reveals a unilateral tumor the involved adrenal should be removed and the patient followed for symptoms and biochemical evidence for involvement of the other adrenal. Bilateral tumors generally indicate the need for bilateral adrenalectomy.
 MCT is the long-term problem since the tumor is frequently aggressive and fatal if untreated. It is preceded by C cell hyperplasia. Any affected individual should have a total thyroidectomy.
MCT is the long-term problem since the tumor is frequently aggressive and fatal if untreated. It is preceded by C cell hyperplasia. Any affected individual should have a total thyroidectomy.
MCT is diagnosed by elevated calcitonin levels. Occasionally, the tumors produce other hormones such as ACTH which results in the ectopic ACTH syndrome. To reiterate, any family member with the trait should have a total thyroidectomy.
 Parathyroid involvement in MEN 2A tends to be diffuse with hyperplasia or multiple adenomas.
Parathyroid involvement in MEN 2A tends to be diffuse with hyperplasia or multiple adenomas.
The clinical manifestations result from hypercalcemia and the complications of hypercalcemia.
MEN 2B
 MEN 2B results from a different mutation in the RET proto-oncogene; it consists of MCT, pheochromocytoma, and mucosal neuromas.
MEN 2B results from a different mutation in the RET proto-oncogene; it consists of MCT, pheochromocytoma, and mucosal neuromas.
Hyperparathyroidism is not present. MCT is more aggressive than in MEN 2A.
 The mucosal neuromas in MEN 2B present as glistening bumps on the eyelids and on hypertrophied lips.
The mucosal neuromas in MEN 2B present as glistening bumps on the eyelids and on hypertrophied lips.
Although the neuromas are presumably present since infancy, MEN 2B frequently escapes detection until early adult life when the syndrome presents with a thyroid mass or paroxysms related to the pheochromocytoma. Management of MCT and pheochromocytoma is the same as for MEN 2A.
 In MEN 2B marfanoid habitus is usually present and ganglioneuromas of the intestine may be associated with bowel motility problems.
In MEN 2B marfanoid habitus is usually present and ganglioneuromas of the intestine may be associated with bowel motility problems.
The causative mutation not infrequently arises de novo so the family history may be negative.
THYROID DISEASE
Thyroid Function Tests
 Thyroid stimulating hormone (TSH) is a reasonable screening test for thyroid disease, but if the clinical suspicion of thyroid disease in an individual patient is high, measurement of circulating thyroid hormones is required as well.
Thyroid stimulating hormone (TSH) is a reasonable screening test for thyroid disease, but if the clinical suspicion of thyroid disease in an individual patient is high, measurement of circulating thyroid hormones is required as well.
When hyperthyroidism is under consideration a total triiodothyronine (T3) level should be obtained in addition to a free thyroxine (T4) level and TSH measurement.
 Normally, 75% to 80% of circulating T3 comes from peripheral conversion of T4; in endogenous hyperthyroidism much more T3 comes from the gland itself as the overactive gland is relatively iodine deficient.
Normally, 75% to 80% of circulating T3 comes from peripheral conversion of T4; in endogenous hyperthyroidism much more T3 comes from the gland itself as the overactive gland is relatively iodine deficient.
As a consequence in hyperthyroidism, particularly Graves’ disease, the T3 level is usually elevated out of proportion to the level of T4.
 A normal T4 level and elevated T3 level has been referred to as “T3 toxicosis.”
A normal T4 level and elevated T3 level has been referred to as “T3 toxicosis.”
 TSH levels are suppressed in all forms of thyrotoxicosis except in the very rare situation of a pituitary adenoma secreting TSH.
TSH levels are suppressed in all forms of thyrotoxicosis except in the very rare situation of a pituitary adenoma secreting TSH.
 Radioactive iodine (RAI) uptake and scan should be obtained in virtually all cases of hyperthyroidism in order to establish the underlying cause.
Radioactive iodine (RAI) uptake and scan should be obtained in virtually all cases of hyperthyroidism in order to establish the underlying cause.
With the exception of the situations described below hyperthyroidism is associated with increased RAI. In Graves’ disease the uptake is diffuse; with a toxic adenoma the uptake is localized to the nodule and suppressed in the remainder of the gland; and in toxic multinodular goiter there are areas of increased uptake interspersed with areas of decreased uptake.
 Hyperthyroidism with a low RAI has a limited and specific differential diagnosis: iodine excess; subacute and postpartum thyroiditis; factitious thyrotoxicosis; and struma ovarii (Table 7-4).
Hyperthyroidism with a low RAI has a limited and specific differential diagnosis: iodine excess; subacute and postpartum thyroiditis; factitious thyrotoxicosis; and struma ovarii (Table 7-4).
Expansion of the endogenous iodine pool occurs with therapeutic doses of sodium iodide or the antiarrhythmic agent amiodarone; the RAI administered to assess uptake is diluted in the greatly expanded pool giving a falsely low calculated uptake of the tracer.
TABLE 7.4 Hyperthyroidism with a Low RAI Uptake

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


